
Abstract
Conventional enzyme-linked immunosorbent assay (ELISA) is a gold standard for screening antibodies and testing for protein or antigen presence. A significant limitation of this assay resides in the fact that only one analyte can be assessed per microplate well. Here, we describe and investigate a new technology consisting of an automated ELISA system in which up to 10 analytes can be measured within one single well, thus improving productivity, accuracy, and repeatability by reducing the amount of human labor required. Another strength of the platform is that a user can load any necessary sets/subsets of beads to perform required assays, with improved flexibility compared to manufactured-loaded arrays for multiplex analysis. We also demonstrate that this system can be used to determine the pathogenicity (i.e., presence of Shiga toxins) and serotype (i.e., Escherichia coli O157) of E. coli isolates.
Conventional enzyme-linked immunosorbent assay (ELISA) is a gold standard for screening antibodies and testing for protein or antigen presence. A significant limitation of this assay resides in the fact that only one analyte can be assessed per microplate well. Here, we describe and investigate a new technology consisting of an automated ELISA system in which up to 10 analytes can be measured within one single well, thus improving productivity, accuracy, and repeatability by reducing the amount of human labor required. Another strength of the platform is that a user can load any necessary sets/subsets of beads to perform required assays, with improved flexibility compared to manufactured-loaded arrays for multiplex analysis. We also demonstrate that this system can be used to determine the pathogenicity (i.e., presence of Shiga toxins) and serotype (i.e., Escherichia coli O157) of E. coli isolates.
Plexarray Format
The system used is based on a novel multiplexing format (U.S. patent pending) developed by Dynex Technologies, Inc. (Chantilly, VA). Undyed polystyrene beads (2 mm diameter) were first coated/treated to enhance adsorption and then stored dry. To build the assay, we passively adsorbed capture antibodies onto the beads (see Immunoassay section below). Then the beads were pressed into cylindrical pockets in the bottom of a custom well to fit into a microplate-sized holder ( Fig. 1 ). This special well-plate was manufactured from black polypropylene and fitted into a rectangular 6 × 9 array in a standard microplate-sized holder ( Fig. 2 ). The construction allowed the rigid polystyrene beads to be forced tightly into the narrower pockets, distending them slightly to form a leak-proof seal. The colorless beads provided a good medium for luminescence, whereas the black color of the plates reduced reflectance and cross-talk between beads sharing the same well. Ten beads were fitted into the bottom of each well. Although the format allows for multiplex assay of 10 different analytes, in this proof-of-principle work, only three beads were loaded with capture antibody in each well ( Fig. 3 ). Each of these three beads was coated with a different antibody. The other seven positions were filled with negative control beads coated with an inert blocker. The identity of the bead coating, and thus the specificity of the assay for each bead, was determined by its physical position, as detailed below.

Cutaway close-up of a single well with beads.

Image of the plate used in the new Dynex system.
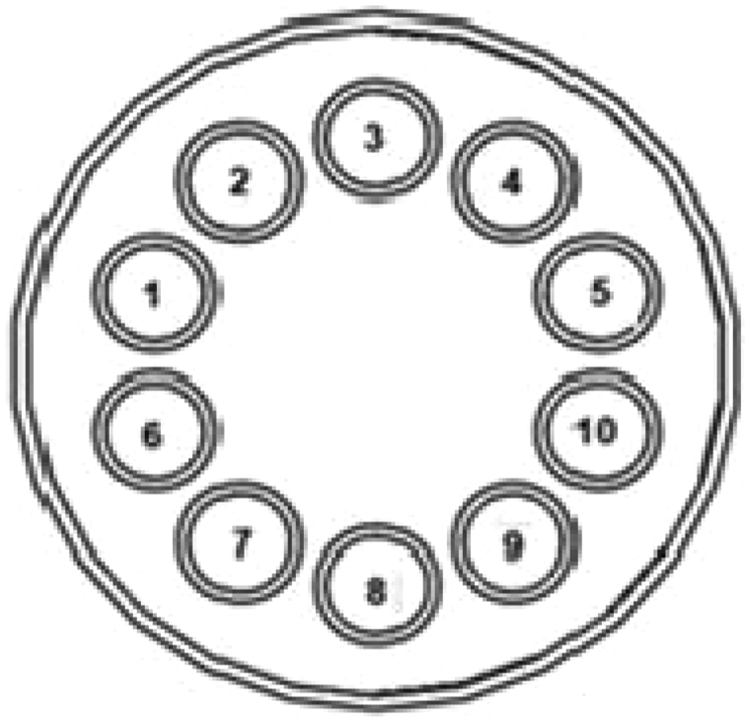
Diagram showing the physical position of the beads used in the current 3-Plex immunoassay. Beads in positions 1, 4, and 9 were coated with anti-Stx1, Stx2, or O157 antibodies, respectively. The remaining beads in positions 2, 3, 5, 6, 7, 8, and 10 were blocked with StabilGuard (SurModics, Eden Prairie, MN).
Liquid Handler
Operation of the assay was greatly facilitated through the use of a liquid handler robot. The Dynex DS-2 Automated ELISA Processing System workstation was used ( Fig. 4 ), which includes all necessary hardware and software for automated sample dilution, reagent addition, temperature-controlled incubation, shaking, and washing of microplate wells. Since 2007, this instrument has been used in clinical diagnostics and other applications, where it is widely used for routine ELISA assays using standard microplate format (not beads). After loading the wash and reagent reservoirs on the DS-2, up to two assay microplates containing beads were placed on the instrument. The instrument was then controlled by computer through a USB serial communication protocol using DS-Matrix software. After the ELISA reaction was completed, the plate was manually removed and mounted into the CCD (charge-coupled device) camera reader.
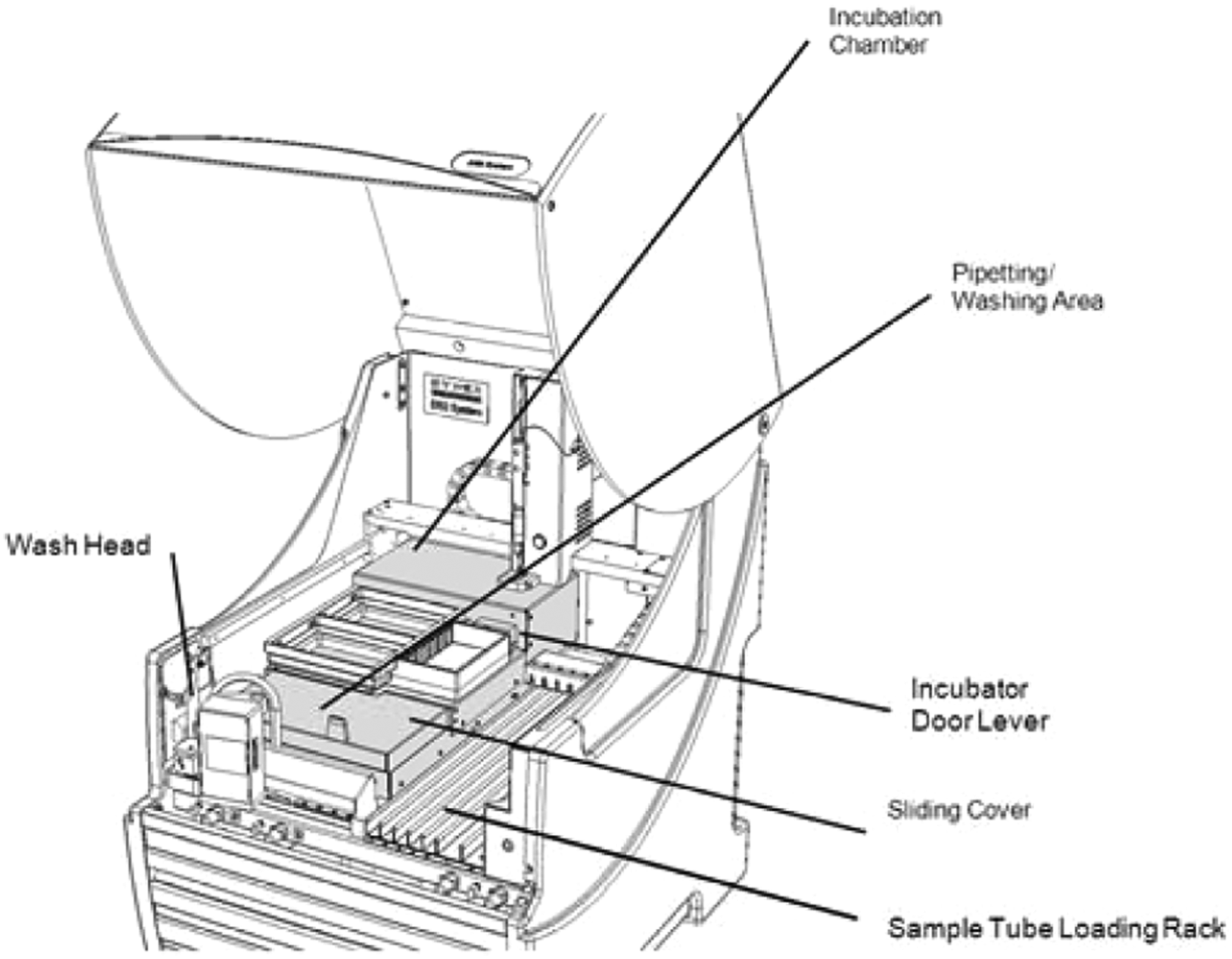
DS-2 Automated ELISA Processing System.
CCD Camera
For chemiluminescent readout of the multiplex assay format, we used a Photometrics (Tucson, AZ) CoolSNAP HQ2 CCD camera ( Fig. 5 ). The camera was mounted within an enclosure designed to exclude extraneous light and maintain proper focus length for detection of light emanating from beads within the microplate. A light-tight door near the bottom of the enclosure allowed insertion of microplates. Microplates were loaded and unloaded manually, one at a time. Images were captured across the entire plate simultaneously, comprising chemiluminescent emission directly related to the presence of analyte. The images were then digitized via Array-Pro Analyzer Software (Media Cybernetics, Bethesda, MD), and the reduction of the raw intensity data was carried out with Microsoft Excel (Microsoft, Redmond, WA).

CCD (charge-coupled device) and camera enclosure.
Macroplexing Beads
The coating of Dynex macroplexing beads is similar to coating a polystyrene microplate with antibodies. In this study, the beads were coated with anti-Stx1 (Shiga toxin 1; Sifin GmbH, Berlin, Germany), anti-Stx2 (Shiga toxin 2; Sifin GmbH), or anti–E. coli O157 lipopolysaccharide (LPS; OEM Concepts, Saco, ME) monoclonal antibodies (mAbs) at a concentration of 1.5 µg/mL. These mAbs were prescreened in an earlier study. 1 The mAbs were prepared in phosphate-buffered saline (PBS, pH 7.4), and the coating volume was typically 50 µL per bead. The beads and coating solution were tumbled slowly on a rotator (Barnstead/Thermolyne, Dubuque, IA) overnight at room temperature (i.e., 18–25 °C). The coating solution was then removed, and the beads were washed three times with PBS-T (PBS, 0.1% Tween-20), blocked in an equal volume (i.e., 50 µL/bead) of StabilGuard (SurModics), and left on a rotator for 2 h at room temperature. The blocking solution was removed, and the beads were then washed with PBS once as before. Beads were then dried at room temperature with vacuum suction on filter paper (Macherey-Nagel, Bethlehem, PA) and stored dry in Sarsted control vials until further use. Coated beads have been found to be stable for >5 months when kept dry at 4 °C with desiccant (Drierite; W. A. Hammond Drierite Co., Xenia, OH).
Bacterial Preparation
A total of 38 E. coli samples were tested ( Table 1 ). Strains were obtained from Drs. Robert Mandrell and Beatriz Quiñones (USDA-ARS, Produce Safety Microbiology Research Unit, Albany, CA) and Andrew Lin (Food and Drug Administration [FDA], SANDO, Alameda, CA). Serotype was determined by Drs. Mandrell and Lin (data not shown), and Stx phenotype was determined by a Luminex sandwich ELISA employing the same mAbs. 1 The E. coli samples were cultured in 5 mL of Brain Heart Infusion broth (BHIB; Oxoid, Cambridge, UK) and incubated overnight (15 h) in a shaking incubator (100 rpm) at 37 °C. Following enrichment, the samples were heat-killed at 55 °C for 2 h. Their optical density at 600 nm was measured, and the samples were diluted to an OD600 of 1. Appropriate BSL-2 (biosafety level) safety precautions were used when handling pathogenic E. coli organisms.
Bacterial Strains Used
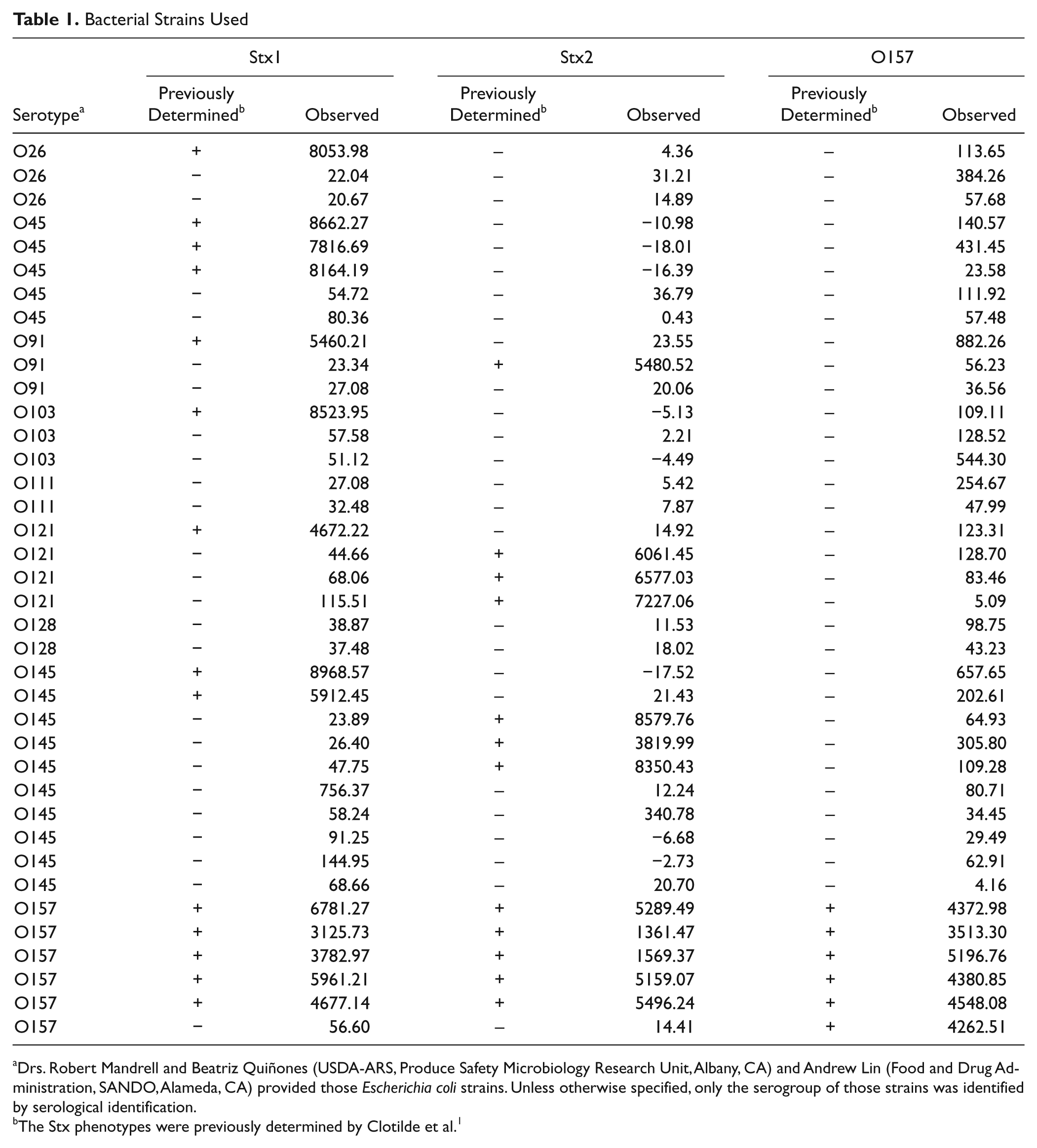
Drs. Robert Mandrell and Beatriz Quiñones (USDA-ARS, Produce Safety Microbiology Research Unit, Albany, CA) and Andrew Lin (Food and Drug Administration, SANDO, Alameda, CA) provided those Escherichia coli strains. Unless otherwise specified, only the serogroup of those strains was identified by serological identification.
The Stx phenotypes were previously determined by Clotilde et al. 1
Immunoassay
Tween-20 was purchased from Sigma Chemicals Co. (St Louis, MO), PBS from Fisher Scientific (Pittsburgh, PA), IgG-free/protease-free bovine serum albumin (BSA) from Jackson ImmunoResearch (West Grove, PA), and streptavidin-horseradish peroxidase (SA-HRP) conjugate from Invitrogen (Carlsbad, CA). The bead-loaded plates were blocked with 500 µL/well of StabilGuard for 30 min at 20 °C with constant shaking. After five washes with 700 µL/well of PBS-T, 300 µL/well of diluted bacterial media was added to the plate and incubated for 30 min at 20 °C with constant shaking. After five washes with 700 µL/well of PBS-T, 300 µL/well of a 0.25-µg/mL solution in StabilGuard of detector mAbs was added to each well and incubated for 30 min at 20 °C with constant shaking. In this assay, the same anti-Stx1 and anti–E. coli O157 LPS mAbs were used for capture and detection, whereas two different mAbs were used for Stx2: Stx2A for capture and Stx2B (Sifin GmbH) for detection. Detector mAbs were biotinylated using the EZ-Link Sulfo-NHS-Long Chain-Biotin kit (Pierce, Rockford, IL), according to the manufacturer’s instructions. After five washes with 700 µL/well of PBS-T, 300 µL/well of a 1/15 000 dilution of SA-HRP conjugate in StabilGuard was added to each well and incubated for 30 min at 20 °C with constant shaking. After five washes with 700 µL/well of PBS-T, 300 µL/well of Luminol solution (Chemiluminescent Super Sensitive HRP Microwell and/or Membrane Substrate; SurModics) was added to each well to detect bound conjugated mAbs. After 2 min of incubation at 20 °C with constant shaking, microplates were manually unloaded from the DS-2 and placed into the CCD enclosure where chemiluminescence was read. The optimal antigen, capture, and detection mAbs concentrations were determined by serial dilutions. For statistical analysis, a signal higher than 1000 was considered positive.
Results and Discussion
The specificity of the mAbs used has been tested previously by microplate sandwich ELISA and microbead-based immunoassay. 1 Our multiplex assay is a qualitative assay constructed around antibody-based detection of the bacterial biomarker antigens O157 LPS, Stx1, and Stx2. With the Dynex macrobeads, as with the previous assay formats, none of the Stx mAbs cross-reacted with any of the 17 non-Stx producing E. coli, nor did Stx1 mAbs cross-react with Shiga toxin-producing E. coli (STEC) that produced only Stx2 or vice versa. Likewise, E. coli O157 LPS mAbs did not cross-react with any of the 32 non-O157 E. coli strains. Non-O157 STEC that we tested belonged to the O26, O45, O91, O103, O111, O121, O128, and O145 serogroups, which make up 74.2% of all non-O157 STEC-related outbreaks of human illnesses in the United States. 2 A corrected signal (signal – background [chemiluminescence emitted by the uncoated beads]) higher than 1000 (arbitrary units) was positive for the analyte tested. Results are shown in Table 1 .
Here we described a new automated platform capable of running an immunoassay testing for the expression Stx as well as the presence of E. coli O157. This platform could easily become a field-deployable advanced assay. It also provides many advantages over conventional sandwich ELISA. First, the Dynex system is a walk-away platform. Second, the Dynex format is more efficient because of its multiplex capability. This reduces sample preparation time and requires less sample material, as well as fewer reagents and consumable supplies. 3 In fact, a conventional sandwich ELISA takes up an entire 8-h day, with about 4 h of incubations and 3 to 4 h of hands-on labor, whereas the Dynex ELISA also takes up a whole day, but the hands-on time is limited to 1 to 2 h of setup and cleanup. A conventional sandwich ELISA requires 100 µL of sample per assay. The Dynex system uses 300 µL for up to 10 assays, which comes to only 30 µL per assay. Although the exact numbers are highly dependent on the specific analytes and assay formats, dramatic improvements in efficiency occur when multiplexing an assay, 4 whereas both speed and reproducibility increase through automated liquid handling. 5 Finally, the greatest strength of this platform is that the user can load the necessary set/subset of beads to perform any required assay, with improved flexibility compared to manufacture-loaded arrays for multiplex. In fact, all existing multiplex ELISA technologies include some kind of array, whether printed spots on microplate wells or fluorescent microbeads manufactured with capture antibody, whereas the Dynex allows the user to choose which assays to include by selecting which macrobeads to fix into the wells.
Our newly developed bead-based immunoassay is capable of detecting Stx1, Stx2, and E. coli O157 LPS simultaneously in growth media. All three multiplexed assays performed with sensitivity and specificity equivalent to previously developed formats. 1 We are encouraged by these preliminary results and are currently working on expanding the assay to include the top six non-O157 antigens and another E. coli virulence factor (i.e., intimin) to fully exploit the capability of 10 simultaneous assays per well.
Footnotes
Acknowledgements
We thank Drs. Robert Mandrell and Beatriz Quiñones (USDA-ARS, Produce Safety Microbiology Research Unit, Albany, CA) and Andrew Lin (FDA, SANDO, Alameda, CA) for providing STEC strains. We also thank Dr. Julian Duncan (London, UK) for his help in data analysis and assay optimization.
Declaration of Conflicting Interests
Coauthors DES, AK, and AF are employees of the Dynex Technologies, currently developing the assay platform described herein as a commercial product. Their role in this work was limited to design and construction of the instrument. Coauthors LMC, CB, and JMC developed and performed the assay and declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Use of trade names and commercial products in this article is solely for the purpose of providing scientific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity employer.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.