
Abstract
The emergence of nucleic acid (NA) therapeutics, including antisense oligonucleotides (ASOs), small interfering RNAs (siRNAs), which are usually delivered directly, and messenger RNAs (mRNAs), which are typically encapsulated in lipid nanoparticles (LNPs), marks a transformative era in precision medicine. While these therapies offer precise approaches for gene regulation or expression, they can trigger unwanted innate and/or adaptive immune responses that can either have no significant impact or adversely affect treatment efficacy and/or patient safety. Consequently, therapies where an adaptive immune response is desired, such mRNA/LNP-based vaccines against infectious diseases or cancer are out of scope of this article. In the present work, the Innovation and Quality Consortium Nucleic Acids Immunogenicity Working Group examines how the various components of NA-based therapies might contribute to their immunogenic potential and describes risk mitigation strategies through product design adaptations during early development stages. In addition, a comprehensive immunogenicity risk assessment framework is described, allowing to effectively define a tailored clinical testing strategy for different NA modalities with varying immunogenicity (IG) consequences. A streamlined monitoring strategy is recommended when minimal impact is expected, whereas extensive testing is suggested when safety concerns arise. Overall, these recommendations ensure that safe and effective NA-based therapies reach patients with an appropriate assessment of the IG potential.
Introduction
Nucleic acid (NA) therapies have achieved remarkable success in recent years demonstrating their potential as innovative treatments for a variety of disorders. 1 Systemic delivery of NAs however is technically challenging due to ubiquitous nucleases present in blood and in cells, which can cleave therapeutic NAs. 2 In addition, mammalian cells possess immune sensing receptors, which can recognize pathogen-derived NAs and potentially trigger inflammatory responses. 3 In recent years, several technological advances such as improved NA design, better chemical modifications, and novel conjugation strategies have effectively addressed these challenges. 4 Furthermore, introduction of nonviral delivery vectors, such as lipid nanoparticles (LNPs), played a pivotal role in the safe and effective transport of NAs to cells in vivo, as evidenced by the approved LNP encapsulated small interfering RNA (siRNA) therapy (Onpattro®). 5
The NA therapies covered in this overview include a variety of modalities such as antisense oligonucleotides (ASOs), siRNAs, and non-vaccine messenger RNA (mRNA). These NA-based gene regulation or protein replacement therapies exhibit different modes of action, but have in common that an immune response is unwanted. Therefore, mRNA/LNP therapies where an immune response is warranted, such as vaccines against infectious diseases or cancer vaccines, are excluded.
ASOs are short, single-stranded sequences of synthetic DNA and/or RNA with 8–35 bases, designed to target specific mRNA molecules and modulate gene expression. ASOs that consist of DNA or RNA/DNA chimeras can induce degradation of the target mRNA via RNAse H. In contrast, RNA-based ASOs bind their target sequence and induce steric blockade, masking specific sequences, and thus modulate splicing. 4 These are referred to as splice-switching oligonucleotides (SSOs). ASOs are usually administered intravenously (IV) or subcutaneously (SC) without encapsulation. They are often covalently linked to a ligand moiety such as the carbohydrate N-acetyl galactosamine (GalNAc) facilitating their uptake into hepatocytes by the asialoglycoprotein receptor. 6
siRNAs typically consist of 21-nucleotide long double-stranded RNA (dsRNA), including an antisense (or guide) strand and a sense (or passenger) strand. The antisense strand binds to the target mRNA sequence via Watson–Crick complementarity leading to recruitment of the RNA-induced silencing complex and subsequent RNA cleavage. 4 Depending on the specific target tissue, siRNAs can be delivered via linking to a ligand or encapsulation in LNPs.
mRNA therapeutics enable transient transgene protein expression with linear mRNA comprising a single-stranded nucleotide chain where the protein encoding sequence is flanked by a 5′ cap structure, 5′ and 3′ untranslated regions, and a 3′ poly A-tail. To prolong the protein expression and potentially reduce the dosing frequency, either circular RNA or self-amplifying RNA (saRNA) can be used. 7 In circular RNA, the ends are covalently fused, which increases its stability. saRNA leverages self-replication from the alphavirus with an RNA-dependent polymerase. 8 Therapeutic mRNAs are often less chemically modified than ASOs or siRNAs to ensure efficient translation. Consequently, mRNAs are more prone to degradation by nucleases in blood and require encapsulation systems such as LNPs for delivery. In addition, LNPs also facilitate receptor-mediated cellular uptake, endosomal escape, and intracellular release of the RNA into the cytoplasm. LNPs typically consist of four components: the ionizable lipid, the polyethylene glycol-conjugated lipid (PEG-lipid), a helper lipid, and cholesterol. While current LNPs enable targeting to the liver, ongoing efforts aim to modulate tissue targeting by altered LNP composition or by incorporating additional targeting moieties. 9
NA-based therapies have a different immunogenicity (IG) profile from traditional small molecules or biologics. Small molecules are generally considered nonimmunogenic, although in rare cases are capable of triggering allergic immune responses. 10 Protein therapeutics on the other hand carry an inherent IG risk and are carefully monitored for their potential to induce (mainly adaptive) immune response, resulting in formation of anti-drug antibodies (ADAs). NA therapies require a new framework for IG evaluation, as both innate and adaptive immune responses may be induced, leading to cytokine release, T cell activation, and antibody (Ab) formation. The efficacy and safety of the various NA modalities can be impacted differently by an immune response. Although IG triggered by siRNAs or gene silencing ASOs without an LNP carrier may not significantly impact therapeutic efficacy and/or safety, it could potentially lead to limited patient acceptance due to infusion- or injection-related reactions. In contrast, a negative outcome on efficacy and/or safety may occur when a therapeutic protein is expressed. Therefore, conducting an immunogenicity risk assessment (IRA) for NA therapeutics is crucial to determine whether a lean or an extensive IG mitigation and monitoring strategy is required to ensure safe and effective NA-based therapies reach patients. In this article, the Nucleic Acids Immunogenicity working group of the Innovation and Quality (IQ) consortium discusses the importance of the IRA in evaluating and mitigating the IG potential of NA therapeutics. The risks associated with various RNA modalities are outlined, along with practical guidance for early de-risking through in vitro testing and product design modifications. A comprehensive approach for developing an IRA-based clinical testing strategy is also provided, along with a road map for presenting these elements in an IRA for regulatory submission.
Unwanted immune responses to NA-based therapies
Innate responses to NA therapies may include hypersensitivity and infusion-related reactions (IRRs) or injection site reactions (ISRs) that manifest with symptoms such as fever, fatigue, or in rare cases, more severe reactions like anaphylaxis. The recognition of NAs is an important mechanism that enables the innate immune system to act as primary defense against microbial infections. This process, known as NA sensing, can be triggered by pattern recognition receptors (PRRs) present both in immune and nonimmune cells. PRRs vary in their cellular localization (endosomal or cytosolic) and in their ligand specificity. Toll‐like receptors (TLRs) 3, 7, 8, 9, and 13 are localized in the endosome and can recognize different structural or sequence patterns in NAs. For instance, TLR3 can bind to dsRNA, 11 whereas TLR7 can be activated by unmodified siRNA. 12 Unmodified single-stranded RNA (ssRNA) can be recognized by TLR7 and TLR8, 13 and bacterial DNA can be sensed by TLR9, either through unmethylated CpG motifs in single-stranded DNA14,15 or double-stranded DNA (dsDNA). 16 In the cytoplasm, important RNA‐specific receptors include DExD/H-box helicases: melanoma differentiation associated protein 5 (MDA5) and retinoic acid-inducible gene I (RIG‐I). 17 While the ability of MDA5 to recognize specific RNA motifs has not yet been characterized in detail, it is known that RIG-I recognizes blunt ends in dsRNA containing a 5′-triphosphate or a 5′-diphosphate.18–20 Binding of stimulatory DNA oligonucleotides to the receptor for advanced glycation end-products (RAGE) on the cell surface facilitates their uptake and subsequent activation of TLR9 downstream signaling. 21 In addition, RAGE activation also increases the sensitivity of all ssRNA-sensing TLRs (TLR7, TLR8, TLR13), suggesting that RAGE is an integral part of the endosomal NA-sensing system. 22 Cytosolic DNA can also be recognized by the cGAS-cGAMP-Stimulator of Interferon Genes (STING) pathway. 23 PRRs activate downstream signaling leading to nuclear translocation of the transcription factor NFκB and a certain subset of interferon regulatory factors which induce the production of cytokines, including interleukin (IL)-1, IL-6, tumor necrosis factor-alpha, and type I interferons (IFNs). 24
Innate immune activation can also provide an adjuvant effect to adaptive immunity. The formation of ADAs against ASOs and siRNA has been reported, 25 and although the precise mechanism is not fully understood, there is evidence that PRRs play a crucial role. For instance, activation of TLRs engages two signaling pathways: the MyD88-dependent and the TRIF-dependent pathway. 26 The MyD88-dependent pathway can activate NF-κB and upregulate several pro-inflammatory cytokines. This signaling pathway can indirectly enhance inflammasome signaling by upregulating pro-IL1β.27,28 It can also have an important role in B cell induction. 29 The TRIF pathway is activated by TLR3 and TLR4 and can promote adaptive immune responses. 30 Furthermore, cytosolic DNA recognition by the cGAS-cGAMP-STING pathway activates expression of Type I IFN (IFNα and β) or IL-1β, 31 although RIG-I and MDA5 can also play an important role in the induction of Type I IFNs. 32 Plasmacytoid dendritic cells are responsible for the production of Type I IFN and are activated by TLR7 and TLR9. 33 Type I IFN can upregulate major histocompatibility complex (MHC) class I in many cell types (including immune cells), with MHC class I being required for cytotoxic CD8 T cell-mediated responses. In contrast, Type I IFN upregulates MHC class II expression on antigen-presenting cells (APCs) leading to activation of CD4 Th cells as part of the humoral immune response. 34 The cGAS sensor can detect longer dsDNA (more than 40 bp) although it can also detect Y-form short dsDNA. 35
Endosomal uptake and escape by membrane disruption is the major mechanism of NA delivery from the extracellular to the cytoplasmic site of action. 36 However, endosomal membrane damage is also a danger signal to the cell resulting in the release of pro-inflammatory cytokines. 37 In addition, the ionizable lipid of the LNP can independently activate the innate immune system resulting in similar clinical symptoms as described for NAs. Historically, cationic lipids have been shown to stimulate TLR2 and TLR4. 24 Empty LNPs without RNA payload can induce IL-1β release, suggesting that lipids alone can activate inflammasomes. 38
Another aspect of innate immune activation relates to the complement system. ASOs with phosphorothioate (PS) and 2′-O-methoxyethyl (2′-MOE) modifications were shown to activate the alternative pathway of the complement cascade.39,40 Complement activation typically occurs within hours after dosing, is dose dependent, and generally transient. Acute or chronic activation, however, can deplete C3, a protein essential for clearing immune complexes (ICs), leading to increased IC deposition in blood vessels, causing vascular inflammation. 41 A recent study by Bakos et al. has shown that LNP-mediated complement activation contributes to the cytokine release observed in human serum in vitro. 42
Another component of the LNP, the PEG lipid, can elicit both innate and adaptive immune responses. Due to the widespread presence of PEG in everyday cosmetic products, the prevalence of preexisting anti-PEG antibodies is high.43,44 At the same time, PEG molecules used in different applications can vary widely in their size, structure (linear or branched), and terminal modifications; thus, the type and impact of preexisting anti-PEG immunity on nucleotide LNPs are not well understood. Anti-PEG Ab formation can also be induced after LNP administration 45 mainly through a T cell independent mechanism, when B cell receptors are directly cross-linked by the repetitive PEG structure. 46 Consequences of anti-PEG antibodies may include accelerated blood clearance (ABC) phenomenon of LNPs through phagocytosis, affecting efficacy as observed with other pegylated biotherapeutics. While the ABC phenomenon has been noted for PEGylated liposomes in mice, 47 it has not been observed in humans for LNPs with rapidly shedding PEG-lipids, even after long-term repeat dosing. 48 Furthermore, anti-PEG antibodies may bind to the PEG on LNPs, potentially triggering complement activation-related pseudoallergic (CARPA) reactions. Although rare, the presence of anti-PEG IgE antibodies may cause anaphylactic reactions. 49
The expressed transgene protein from mRNA/LNP or SSOs therapies carries the potential to elicit adaptive immune activation, including T cell response and induction of antitransgene protein antibodies similar to ADAs observed with biotherapeutics. For simplicity, the term ADA is used in the article to also refer to these anti-transgene antibodies. Furthermore, any peptide or protein-based targeting moiety used to enable targeted LNP delivery may also induce ADA formation. The amino acid sequence of these proteins significantly contributes to the IG risk and ADA development, a concept that has been extensively studied for recombinant protein therapeutics and is also applicable here. Key intrinsic feature of a given protein is the presence of T cell epitopes in the primary amino acid sequence, with a potential to induce activation of CD4 T cells by HLA II-restricted epitope presentation followed by production of ADA. 50 In addition, since the mRNA-encoded transgene protein will be initially expressed intracellularly, HLA I-restricted epitopes might also be generated. These can potentially activate cytotoxic CD8+ T cells, which in turn can lead to cell killing, loss of treatment efficacy, and tissue damage. 51 The transgenic protein can be secreted, membrane bound, or expressed intracellularly. Accordingly, the risk of ADA development and the potential impact of the ADA can vary. ADAs can be classified either as binding or neutralizing and may adversely affect the transgenic protein exposure and/or function. If the transgenic protein has an endogenous counterpart, and neutralizing ADAs (NAbs) are formed that cross-react with an endogenous protein, they may lead to an autoimmune phenotype that compromises patient safety. In contrast, for intracellularly expressed transgene proteins the formation of ADAs is possible but less concerning since no impact is expected on transgenic protein level or efficacy. In this case, cellular responses mediated by cytotoxic CD8+ T cells are of greater concern. 52 Similarly, consideration to cytotoxic T cell responses should also be given, when saRNA is used, as viral RNA polymerase is expressed intracellularly (for RNA self-amplification) that could at least theoretically be presented on MHC-I. 53 In addition to the treatment-emergent adaptive immunity, the presence of preexisting ADAs (PE-ADAs) has been reported for therapeutic proteins 54 and should be considered for any protein moiety of the nucleotide therapy.
Clinical observations of marketed products
We conducted a comprehensive analysis of nonclinical and clinical innate and adaptive IG events observed for NA therapeutics and summarized them in Supplementary Table S1 and Table 1, respectively. Among the six ASOs, three were administered SC every week at doses of 200–300 mg per patient (3–4 mg/kg). High ADA incidence (30%–70%) with a broad titer range and mild-to-moderate ISRs (50%–99%) were observed, along with complement factor C3 consumption. A GalNAc-ASO administered at 0.6 mg/kg SC monthly resulted in a lower ADA incidence (37%) and ISR rate (7%), suggesting that lower doses and frequencies reduce ISR but not ADA incidence. Intrathecal (IT) administration at a 10-fold lower dose showed a similar trend. In general, ADA formation caused decreased plasma clearance and increased trough levels (Ctrough) without significantly affecting peak plasma concentration (Cmax) or total area under the curve, the latter determined in the first 24 hours after administration reflecting the limited clinical impact of ADAs against NAs. Model-based approaches, population pharmacokinetics (PK) and population PK/pharmacodynamic (PD) modeling, can be used to quantify the IG impact on exposure, efficacy, and safety. Adverse events, including complement consumption and platelet reduction, were primarily associated with weekly administrations of higher dose ASOs.
Overview of Clinical Immunogenicity for Marketed Nucleic Acid Products

2′-F, 2′-fluoro; 2′-MOE, 2′-O-methoxyethyl; 2′-OH, 2′ hydroxyl; 2′-OMe, 2′-O-methyl; ADA, anti-drug antibody; ALS, amyotrophic lateral sclerosis; ASCVD, arteriosclerotic cardiovascular diseases; ASOs, antisense oligonucleotides; C3, complement factor 3; CFTR, cystic fibrosis transmembrane conductance regulator; Dlin-MC3-DMA, dilinoleyl-methyl-4-dimethylaminobutyrate; DMG-PEG2000, 1;2-dimyristoyl-sn—glycero-3-methoxypolyethylene glycol; DOPE, dioleoyl phosphatidylethanolamine; DSPC, 1;2-distearoyl-sn-glycero-3-phosphocholine (INN: Colfoscerilstearat); FCS, familial chylomicronemia syndrome; hATTR-PN, hereditary transthyretin amyloidosis with polyneuropathy; HeFH, heterozygous familial hypercholesterolemia; ICE, imidazole cholesterol ester; IFN, interferon; IL, interleukin; IRR, infusion-related reaction; ISF, infusion site findings; ISR, injection site reaction; IT, intrathecal; LDL-C, low density lipoprotein-cholesterin; LLN, lower limit of normal; DMD, Duchenne muscular dystrophy; MCP-1, monocyte chemoattractant protein-1; MIP-1α, macrophage inflammatory protein-1 alpha; n.e., not evaluated; PA, propionic acidemia; PCC, propionyl-CoA carboxylase (PCC) composed of six alpha (PCCA) and six beta (PCCB) subunits; PCSK9, proprotein convertase subtilisin/kexin type 9; PD, pharmacodynamic; PEG2000-C-DMG, (R)-methoxy-PEG2000-carbamoyl-di-O-myristyl-sn-glyceride; PH1, primary hyperoxaluria type 1; PK, pharmacokinetic; PO, phosphodiester; PMO, phosphorodiamidate morpholino oligonucleotides; SC, subcutaneous; SMA, spinal muscular atrophy; SSO, splice-switching oligonucleotide.
Phosphorodiamidate morpholino oligomer (PMO) SSOs dosed weekly IV (30–80 mg/kg) demonstrated markedly lower IG, with rare and mild ISRs, and no ADAs. One SSO product triggered anti-transgene protein antibodies in 3.13% of patients, but without impact on the intracellular transgene protein level. For the other SSOs (three expressing the same and one expressing another intracellular protein), ADA formation was either not investigated or not detected.
GalNAc-siRNAs (doses: 2.5–284 mg/kg administered SC monthly or <10 mg/kg administered every 6 months) had low ADA incidences (1%–6%), with mild-to-moderate ISRs (4.1%−32.5%), typically resolving in 3–6 days. Notably, longer dosing intervals (every 3 or 6 months) resulted in reduced ISR incidences (<10%), despite the administration of higher doses. Importantly, ADAs did not affect PK or treatment efficacy.
For siRNA/LNP (0.3 mg/kg, IV), low and transient anti-PEG ADAs (3.6%) were observed which did not affect PK, efficacy, or safety. It remains uncertain whether the rapidly shed low-molecular weight PEG in LNPs may lead to adverse events. 88 Although elevated anti-PEG Ab levels were noted after Comirnaty and SpikeVax administration, 45 no significant impact was observed and anaphylaxis rates remained low. 89 No significant anti-PEG Ab formation has also been reported for other LNPs in development.48,90 Despite these uncertainties, vigilance and monitoring for anti-PEG antibodies remain important and expected by health agencies (HAs).
Overall, ADA formation against NAs remained without clinical significance, while dose reduction and less frequent administration significantly reduced, but did not prevent, the ISR rates which lead to treatment discontinuation in early marketed products. Given the very limited clinical experience with SSOs that target only two different transgenes and the absence of approved nonvaccine mRNA therapies, minimal IG impact on the transgene protein has been observed so far. However, the considerable experience within the biotherapeutics field provides valuable insights that underline the need for caution, careful risk evaluation, mitigation, and monitoring.
Immunogenicity risk assessment
The concept of the IRA was initially established for biotherapeutics91–95 and has evolved to cover new modalities, including NA therapies. 96 It comprises three key steps as follows: identifying risk factors, evaluating their potential impact on efficacy and/or safety, and defining appropriate risk monitoring and mitigation strategies (Fig. 1). Initiating the IRA early and revising throughout development by a multidisciplinary team, as recommended by regulatory agencies, including the FDA and EMA,91,92,97 the IRA facilitates rational product design and informed risk management. It enables tailored monitoring strategies in nonclinical and clinical studies and the development of risk mitigation plans if necessary. Furthermore, the IRA justifies a more streamlined monitoring approach for therapies with limited IG impact or, alternatively, highlights the need for more in-depth monitoring to appropriately address risk and indicates whether regulatory consultation might be needed. The IRA differs from the overall benefit–risk assessment, which provides a broader perspective by weighing the overall therapeutic benefits against all associated risks, including but not limited to IG, while considering the severity of the disease and the availability of alternative treatment options. For example, in life-threatening conditions with limited or no alternative treatment options, there may be greater acceptance of therapy-associated risks, including IG, requiring additional management efforts. Conversely, treatment discontinuation might be more readily considered in nonlife-threatening situations where an IG impact on efficacy and safety (incl. tolerability) is observed. The IRA should be included in the Integrated Summary of Immunogenicity, which consolidates nonclinical and clinical IG data submitted to HAs during the marketing application (Fig. 2). In addition, according to recommendations from the FDA, the IRA should be provided along with the rationale for the IG testing paradigm in the IND application to gain alignment for the clinical mitigation and monitoring strategy. 97 The recent FDA guidance on oligonucleotides emphasizes assessing IG for all structural components, including the carrier (e.g., PEGylated lipid), backbone (with modifications), and oligonucleotide sequence along with any novel epitopes created by the drug. It suggests implementing a multitiered IG testing strategy for humoral responses and, importantly, to consider measuring innate immune system activation where appropriate. 98 These recommendations are further supplemented by recent white papers.41,99,100
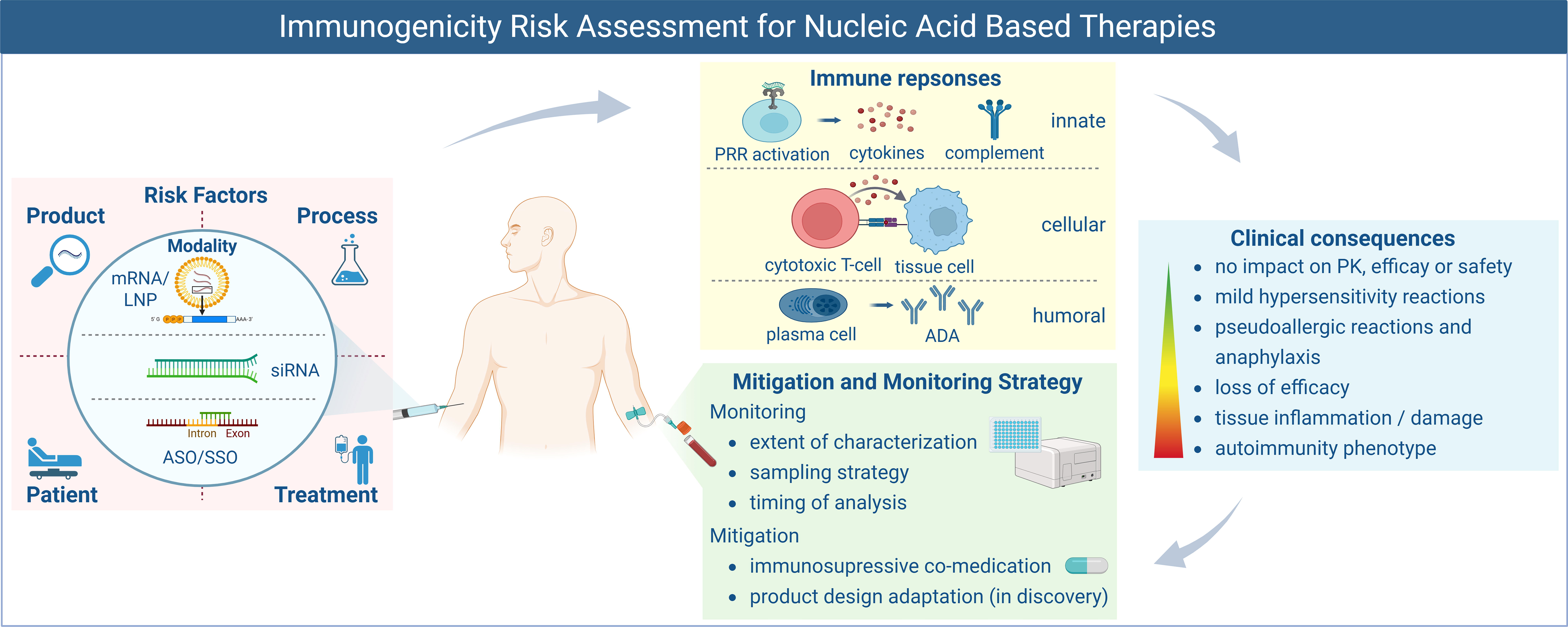
The immunogenicity risk assessment process consists of risk factor identification, evaluation of potentially elicited immune responses, and their clinical consequences facilitating the definition of a tailored mitigation and monitoring strategy.
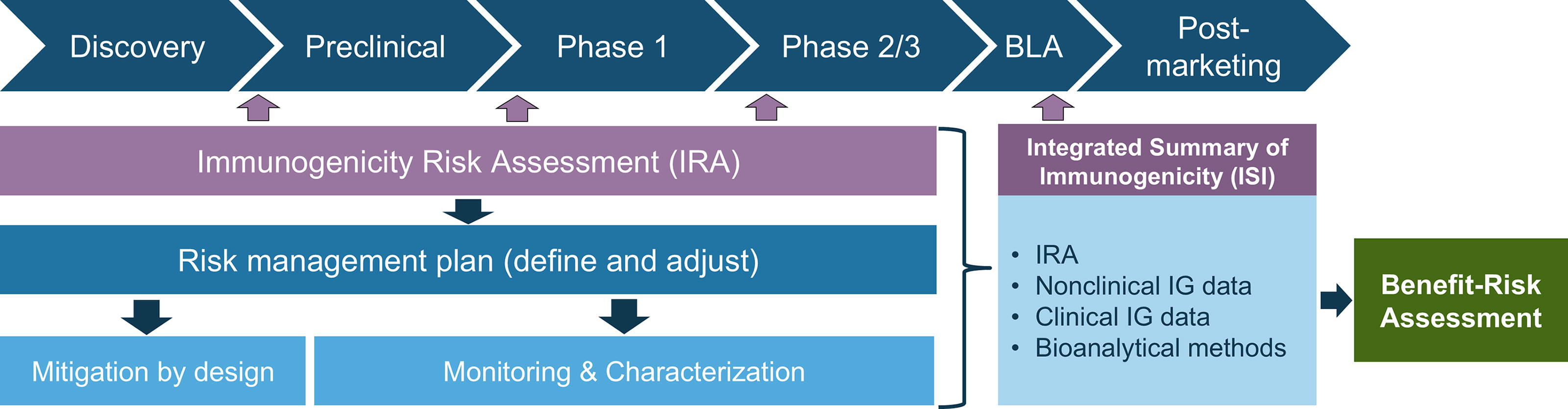
The immunogenicity risk assessment impact during drug development which is part of the integrated summary of immunogenicity submitted to health agencies upon market authorization. The immunogenicity risk assessment is also part of the benefit–risk assessment.
The IRA starts with the identification of all potential IG risk factors associated with the NA therapy components followed by evaluating their potential to trigger immune activation and impact on efficacy and/or safety (Fig. 1). In the absence of clinical data, insights from similar therapies, prior projects, and literature can inform the assessment. Similar to other modalities, IG risk factors can be grouped into product-, process-, patient-, and treatment-related categories (Table 2). Product-related risks include the NA type (ASO, siRNA; linear, circular, or self-amplifying mRNA), its base content, nucleoside modifications, conjugations (e.g., GalNAc), and presence of dsRNA (capable of inducing innate response). For LNP formulations, key concerns include the choice of ionizable lipid (potential innate immune activation), presence of PEG, and/or the use of protein-based targeting moieties (the latter two potentially inducing ADA formation). For mRNA-based therapies and for SSOs, the IG risk related to the transgene protein expression must also be considered, including the protein sequence origin, its homology to endogenous counterparts, and the protein expression pattern (secreted, intracellular, or transmembrane), to assess whether ADA formation and/or cytotoxic T cell responses are of concern.
Overview of Immunogenicity Risk Factors for Nucleic Acid Therapies
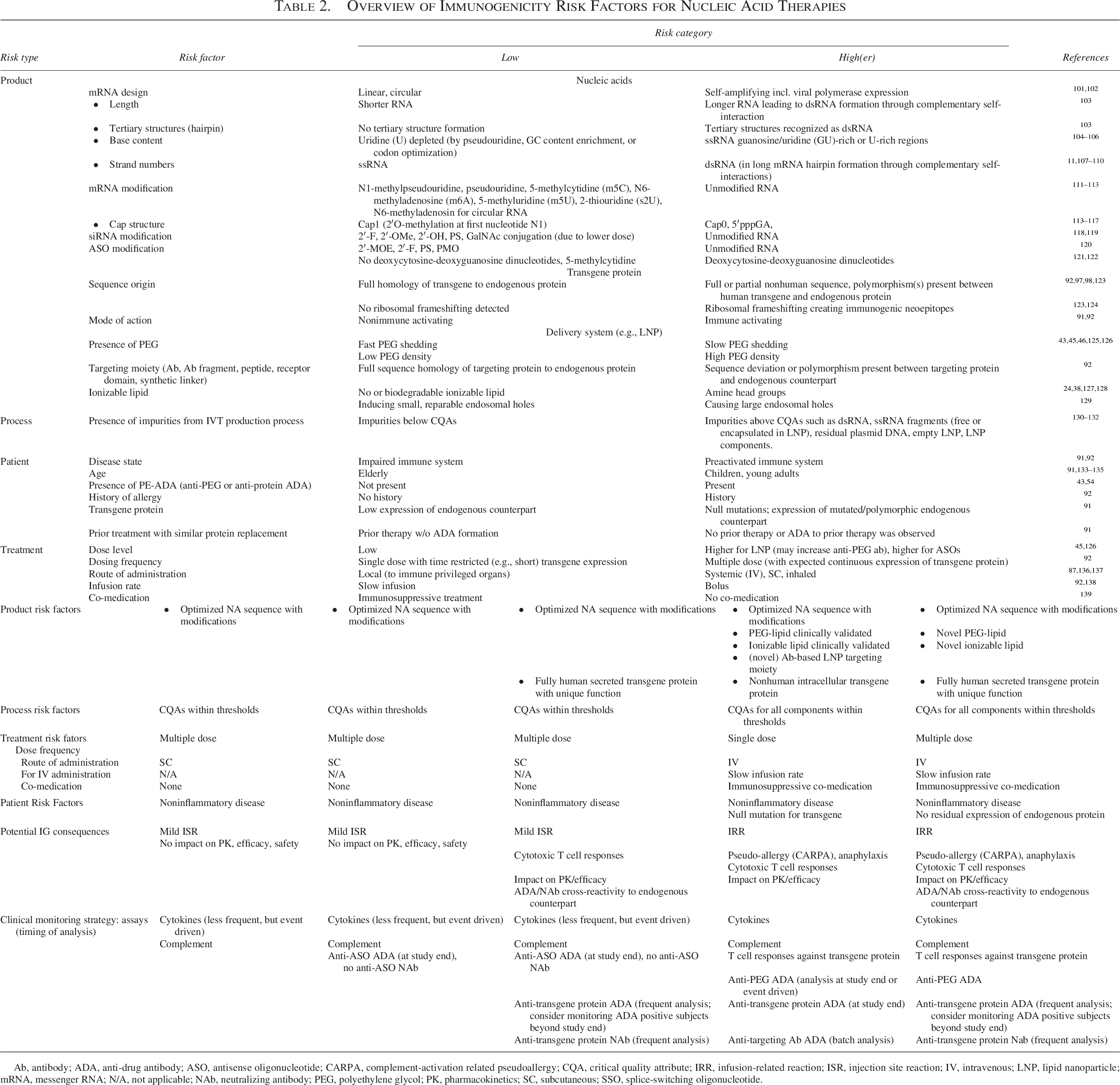
2′-F, 2′-fluoro; 2′-MOE, 2′-O-methoxyethyl; 2′-OH, 2′ hydroxyl; 2′-OMe, 2′-O-methyl; Ab, antibody; ADA, anti-drug antibody; ASO, antisense oligonucleotide; bp, base pair; DNA, deoxyribonucleic acid; dsRNA, double-stranded ribonucleic acid; GalNAc, N-acetyl galactosamine; IV, intravenous; IVT, in vitro transcription; LNP, lipid nanoparticle; mRNA, messenger ribonucleic acid; PE-ADA, preexisting ADA; PEG, polyethylene glycol; PMO, phosphorodiamidate morpholino oligonucleotides; pppGA, guanosine triphosphate; PS, phosphorothioate; SC, subcutaneous; siRNA, small interfering ribonucleic acid; ssRNA, single-stranded ribonucleic acid.
Process-related risks arise from impurities introduced during manufacturing, including those from the LNP delivery system or the presence of endotoxins. 140 For ASOs and siRNAs, which are synthetically produced and relatively short in size, structurally related or unrelated truncated products or the presence of starting material-derived impurities should be evaluated to understand the IG potential.130,141 In contrast, mRNAs are generated through in vitro transcription, are longer in size, and are delivered via LNPs, therefore often have a more complex impurity profile such as residual plasmid DNA templates, enzymes, free nucleotides, dsRNA, incomplete mRNA synthesis products (including uncapped RNA) as well as empty or impurity-loaded LNPs.131,132 These contaminants can trigger innate immune responses, and therefore, several techniques have been developed to detect, avoid, or remove them.142–145 When assessing the transgene, on the upside, unlike biotherapeutics produced in cell lines incorporating nonhuman post-translational modifications (PTMs) contributing to the IG risk, mRNA-expressed transgene proteins possess endogenous PTMs, which may lower the IG risk.
Patient-related risks of NA therapies are similar to those of biotherapeutics and encompass the disease state, the immune status (autoimmune, inflammatory, or immunosuppressed), and previous treatment(s). In addition, PE-ADAs, such as against the PEG, the transgene, or the NAs need to also be considered as they may reduce therapeutic efficacy and increase the likelihood of adverse immune reactions. 92 Evaluating the protein component (e.g., the transgene protein or a protein targeting moiety within the LNP) necessitates assessing the patient’s genetic background and the degree of immune tolerance to the endogenous protein, which often depends on its abundance in the relevant patient population. 92 Full sequence homology with the endogenous counterpart generally lowers the risk of triggering an adaptive immune response (i.e., ADA formation and T cell activation) while sequence deviation(s) or polymorphisms can raise it. Safety concerns arise when ADAs target the protein moiety. Functional neutralization of the endogenous protein by cross-reactive ADAs can induce an autoimmune phenotype, worsening the patient’s condition, especially if the endogenous protein has a unique role. Patients with null mutations for the transgene protein are more likely to mount adaptive responses, potentially compromising therapeutic success. This risk may be lower if prior treatment with a homologous protein replacement therapy did not elicit ADA formation. 91
Treatment-related risks vary between single dose and chronic treatment, as IG elicited by a one-time therapy will likely have reduced consequences on efficacy and safety compared with chronic use and will require different mitigation and monitoring strategies. The route of administration can also influence the IG risk profile. For example, no innate activation was observed upon direct myocardial injection of mRNA. 146 For systemic administration, immunosuppressive co-medication may further reduce the risk.
Overall, the IG profile of NA therapies depends on multiple factors, affecting innate, cellular, and humoral immunity. The consequences may range from negligible impact on PK, PD/efficacy, or safety to mild IRR or even severe hypersensitivity responses such as pseudoallergy or anaphylaxis. Potential IG concerns for therapies involving transgene expression or targeting proteins can include T cell mediated tissue inflammation or damage,147,148 loss of efficacy (which is a safety concern in life-threatening diseases), and the potential development of an autoimmune phenotype.
Immunogenicity risk mitigation and management strategies
After a thorough evaluation of all IG risk factors, a risk management plan is established. De-risking activities vary, depending on the project stage. In early research, before candidate selection, the IRA focuses on the evaluation of product-related risks, including the molecular design and the mode of action. At this stage, only certain aspects of patient and treatment-related risks (e.g., the disease or immune state and the anticipated treatment regimen) can be considered, whereas process-related risks cannot be assessed. However, conducting the IRA in early research enables preventive mitigation by altering the product design (e.g., introducing chemical modifications) and selecting a lead candidate with the lowest IG risk. Further de-risking may include targeted delivery, for example, by utilizing delivery systems that direct NA therapies to specific tissues or cells, which can minimize systemic exposure and widespread immune activation. In addition, careful patient selection based on the immune status and potential preexisting conditions in the population that may predispose individuals to an immune response should also be considered. Before clinical development, the IRA expands to cover all IG risk categories (product-, process-, patient-, and treatment-related risks) and informs both the analytical monitoring strategy to allow for a thorough evaluation and characterization of actual IG in patients and the implementation of additional de-risking measures, including immunosuppressive co-medication.
Optimizing the design of NA therapies
When developing therapeutic NA products, various design strategies can be implemented to mitigate IG. In ASOs, multiple factors such as structural motifs, sequence alterations, and incorporation of chemical modifications help to reduce the IG risk and improve stability, safety, and efficacy. 41 Modifications can be introduced at the phosphate group such as PS, methylphosphonate, and phosphoroamidate or at the sugar moiety, including 2′-O-Methyl (2′-OMe) and 2′-MOE as well as locked NAs (LNAs). Whole backbone modifications, including peptide NA and PMOs, can also be used. 120 Modifying bases or backbone such as avoiding deoxycytosine-deoxyguanosine dinucleotides can reduce the immunostimulatory properties of CpG motifs, thereby mitigating TLR9 activation.121,122 Alternatively, if a CpG motif cannot be avoided, the replacement of the cytidine nucleotide with a 5-methylcytidine can mitigate CpG-dependent immunostimulation. 65 Common ASO engineering strategies also include a gapmer-based design where a DNA core is flanked by RNA wings, including LNA nucleosides, to improve affinity for the target RNA sequence. However, it has been shown that LNA modification does not prevent TLR9 activation. 149
Single-stranded PS backbone containing oligonucleotides can activate the complement system via direct binding to complement factor H, a regulatory protein that plays a role in the complement system or by binding to plasma proteins triggering the alternative pathway. To address these concerns, it is recommended to screen for complement activation by conducting in vitro assays before clinical advancement. 150 In addition, the risk of complement activation can be reduced by using slow or continuous IV infusion, which substantially lowers the maximum plasma concentration compared with bolus injection. 138
Naked and unmodified siRNAs can activate TLR3119,151,152 although this can be dependent on the length of the dsRNA. 153 TLR7/8 can also be activated by unmodified blunt-ended siRNAs. 118 There are strategies to mitigate an innate immune response by the insertion of chemical modifications, including replacing the sugar 2′ hydroxyl group with 2′-Fluoro or 2′-OMe. 118 PS bonds commonly used in ASOs enhance metabolic stability, but are less prevalent in siRNAs. 154
Key approaches in mRNA optimization include codon optimization, chemical modification of nucleosides, the choice of the Cap structure as well as the utilization of appropriate delivery vehicles that encapsulate the therapeutic NA. Karikó and Weissman discovered that substituting uridines with pseudouridines can circumvent the inflammatory effects of mRNA in vivo. 111 Other studies also showed that uridine repeats may trigger inflammation through recognition by TLR7 and TLR8.13,155,156 One mitigation strategy involves using codon optimization to replace uridines without altering the amino acid sequence.104,157 However, this can result in increased GC content, which may potentially lead to misfolding of mRNA. 158 Alternatively, substituting uridines with pseudouridines or N1-methylpseudouridines, which are naturally occurring RNA modifications, preserves mRNA-ability to encode proteins accurately, 159 enhances protein expression, and suppresses immune activation by avoiding RNA sensors, such as 2′−5′-oligoadenylate synthetase, protein kinase R, and RNase L.160–164 In addition, mRNA circularization significantly reduces RIG-I and TLR recognition without the need for chemical substitution. 101 However, N1-methylpseudouridine modification can cause low-level ribosomal frameshifting (8%), 123 a natural mechanism in which the ribosome can slip into a +1 or −1 alternative reading frame thus changing the downstream amino acid sequence, 124 potentially creating neoepitopes that could trigger an adaptive immune response.165–167 Although no off-target safety concerns have been attributed to current mRNA-LNP vaccines, 123 it is a factor to consider. Sequence optimization at frameshift-prone “slippery sites” may mitigate this phenomenon. 168 A recently developed liquid chromatography–tandem mass spectrometry (LC-MS/MS)-based assay enables characterization of mRNA translation fidelity in vaccines. 165
LNP design optimization can also play an important role in the IG risk mitigation strategy of NA drugs requiring encapsulation. A significant improvement was achieved after switching from cationic to ionizable lipids. 169 Further improvements could be achieved by modifying the LNP composition such as altering size, molar ratios, PEG component ratio, and formulation rate and by adjusting factors during the LNP formulation process. 46 To mitigate the risk of inflammation associated with endosomal disruption caused by the LNP, it is crucial to focus on the size and reparability of endosomal holes. Smaller, reparable holes lead to reduced inflammation and higher mRNA expression, whereas larger, irreparable holes may induce innate activation. 129 Galectins, which bind to sugars on the inner endosomal membrane, recognize this damage and trigger inflammation. Inhibiting galectins can prevent LNP-associated inflammation. In addition, using biodegradable ionizable lipids can create smaller, repairable endosomal holes through the endosomal sorting complex required for transport pathway, resulting in high RNA expression with minimal inflammation. 129
Further LNP optimization can include PEG substitution with biodegradable polymers (e.g., polysarcosine) or changing the length or molecular weight of PEG lipids. 46 Anti-PEG antibodies have in very rare cases caused (severe) allergic reactions to PEG-containing LNP. 43 Therefore, it has been suggested that LNPs with PEG substitution should be tested for allergenicity potential using in vitro assays that measure the risk of mast cell degranulation and basophil activation. 89 Alternatively, fast PEG shedding (e.g., 2-dimyristoyl-rac-glycero-3-methylpolyoxyethylene-2000 (PEG2000-DMG)) reduces anti-PEG IgM response compared with the slower shedded 1,2-distearoyl-rac-glycero-3-methylpolyoxyethylene-2000 (PEG-DSG) as shown in mice. 125 Fast PEG shedding of up to 45% per hour was demonstrated for PEG2000C-DMG in mice, 170 which is also included in Onpattro® where low incidence of transient anti-PEG antibodies has been reported in human without impacting PK and PD/efficacy or safety. 171
Mitigation of IG risk related to the transgene protein can include the identification, characterization, and removal of CD4+ associated HLA II- and CD8+ T associated HLA I-restricted epitopes. In silico and in vitro tools, including epitope prediction algorithms, are used to identify T cell epitopes based on HLA binding affinity.172,173 However, such algorithms do not inform the epitope T cell activation risk. This can be predicted using algorithms that estimate foreignness: 174 the more foreign to the human immune system, the more likely an epitope will induce a T cell response. In vitro studies, such as APC activation and T cell activation assessments, may further evaluate the likelihood of epitope presentation. 175 However, removing deleterious T cell epitopes may impact the transgene’s paramount properties (e.g., target binding affinity, target specificity) and lead to decrease or even loss of activity. 176 Also worth mentioning that removal of T cell epitopes may potentially create B cell epitopes, 177 which can be interrogated by in silico B cell epitope prediction tools. 178 Advances in in vitro and ex vivo models, such as induced pluripotent stem cell-derived organoids and organs-on-chips, present valuable opportunities for mechanistic investigations, ultimately aiding in the engineering of safer NA-based drugs. However, it is important to recognize that analyzing specific aspects through in vitro assays can be challenging, as the human IG profile in vivo may be more complex than the sum of its individual components.
Immunogenicity analysis during nonclinical and clinical development
Appropriate monitoring of innate and adaptive immune responses is essential to understand the IG profile of NA therapies and its impact on treatment efficacy and patient safety. Each therapy type presents unique challenges, but several common and specialized approaches are used to comprehensively assess immune activation.
In nonclinical and clinical studies cytokine and chemokine profiling is conducted with multiplex bead-based assays [e.g., electrochemiluminescence (ECL) or Luminex] to assess pro-inflammatory and anti-inflammatory markers. Innate immune receptor activation (e.g., TLR7 and TLR8) is analyzed in isolated PBMCs or whole blood by cytokine and chemokine release and gene expression profiling in stimulated immune cells. 183 Complement activation is monitored by measuring components such as C3a, Bb, and -C5b-9 in serum. 39 Cell activation and proliferation can be monitored by flow cytometry using specific markers of the different cell types. 184
Additional analysis not routinely required may include transcriptomics and proteomics analyzing gene and protein expression changes in treated animal tissues and profile patient-specific immune signatures to identify immune-related pathways, explore innate immune activation pathways, and identify predictive biomarkers for patient stratification and therapeutic outcome.
Key challenges include differentiating immune responses against the NA (components) versus the LNP carrier and ensuring assay sensitivity for low-frequency responses.
Histopathology evaluates tissue samples for immune cell infiltration, inflammation, and immune activation in animal studies, with rare application to biopsy samples for assessing immune-related adverse events in humans.
Positive control (PC) antibodies are essential for ADA assay development. For NAs, these are generated through immunization with the NA conjugated to immunogenic carrier proteins (e.g., Keyhole limpet hemocyanin) alongside adjuvants or with phage display. While this approach frequently produces effective PCs, NAs with diminished immunogenic properties may require chemical and/or structural modifications (e.g., blunt ends with a 5′-triphosphate or prolonged use of PS modifications) to enhance their immunogenic potential. 185 Availability of recombinant transgene proteins in mRNA/LNP or SSO therapies and differences in PTMs may pose a challenge for PC generation and ADA method development. In addition, differences in PTMs can complicate ADA detection. In rare cases, isotyping with subclass determination for IgM or IgE-specific ADAs may be needed in cases of hypersensitivity (e.g., urticaria or infusion reactions) requiring isotype-specific PCs.
Assays detecting anti-PEG antibodies face challenges with PC availability and complexities in method setup. 43 Using the entire LNP instead could theoretically facilitate ADA detection against the protein corona—a protein layer on the LNP surface. 186 However, the protein corona components should be immune-tolerant and their in vitro mimicking is challenging, providing no significant added value in light of the currently low impact of anti-PEG ADAs observed.
ADA method validation should follow regulatory guidelines,91,97 with statistical cut points established to differentiate between positive and negative samples in the relevant patient population.187,188 Determining cut points for preexisting ADAs requires stringent assay conditions, specificity checks, and possibly further sample purification. 189 Furthermore, demonstrating specificity in ADA and NAb assays for transgene proteins that share homology with an endogenous counterpart may not always be feasible.
In clinical studies, a multitiered testing strategy that comprised screening, confirmation, and titration is used, 97 whereas in nonclinical settings a lean validation or qualification and only a screening ADA assay suffice when in line with the IRA.187,188,190 ADA assays must distinguish between preexisting, treatment-boosted, and treatment-emergent responses. 191 In summary, immune response monitoring for NA therapies involves complex assays tailored to molecular structures and immune mechanisms with careful selection of assay formats, PCs, and profiling for accurate, reproducible, and clinically relevant IG data.
Clinical mitigation of immune responses
During clinical development of LNP-encapsulated siRNA and mRNA therapies, IRRs were managed with premedication protocols, including oral administration of acetaminophen or ibuprofen, IV administered corticosteroids (e.g., dexamethasone 10 mg or equivalent) as well as H1 (e.g., diphenhydramine 50 mg or equivalent) and H2 (e.g., ranitidine 50 mg or equivalent) blockers before drug infusion.48,86,192,193 In ASOs and siRNA-based therapies without encapsulation, no specific intervention was reported to manage ISR. In severe adverse events, treatment discontinuation should be considered. However, in case of life-threatening diseases where no alternative medication is available, prolonged immunosuppressive co-medication may be considered.
IQ recommendations on IRA-based clinical monitoring strategy
It is recommended to conduct the IRA before clinical development to establish a monitoring strategy for innate, cellular, and humoral immune responses. The IRA guides the assay strategy, sampling times, and the extent and timing of the analysis. Pre- and post-dose IG samples should be collected coinciding with PK and PD time points, with ad hoc sampling allowed in the study protocol in case of adverse events. Innate and cellular responses are typically characterized promptly after collection when assays require PBMC isolation from fresh blood. However, cytokine induction assays, which can be conducted using frozen serum, can be postponed to later phases of the study provided that no safety concerns arise requiring a timely analysis. The timing of ADA analysis should be driven by safety concerns. For single-dose treatments, ADA analysis may be delayed if no safety issues are anticipated or observed. In contrast, immediate analysis, including neutralizing anti-transgene Ab assays, may be more appropriate for chronic treatments, especially when the transgenic protein is replacing an endogenous counterpart with a unique function. For programs with significant safety concerns regarding ADA formation against the protein, it may be necessary to monitor ADA-positive subjects beyond the study completion.97,194 Table 3 outlines how the IRA directly informs the complexity and extent of monitoring required for the different theoretical examples of NA-based therapies. Note that a tailored monitoring strategy needs to be adapted to the specific characteristics of a NA therapy, and it is recommended to align this strategy with health authorities before start of clinical development.
Examples on Different Theoretical Nucleic Acid Therapies with Recommendations for an Immunogenicity Risk Assessment-Based Monitoring in Clinical Development
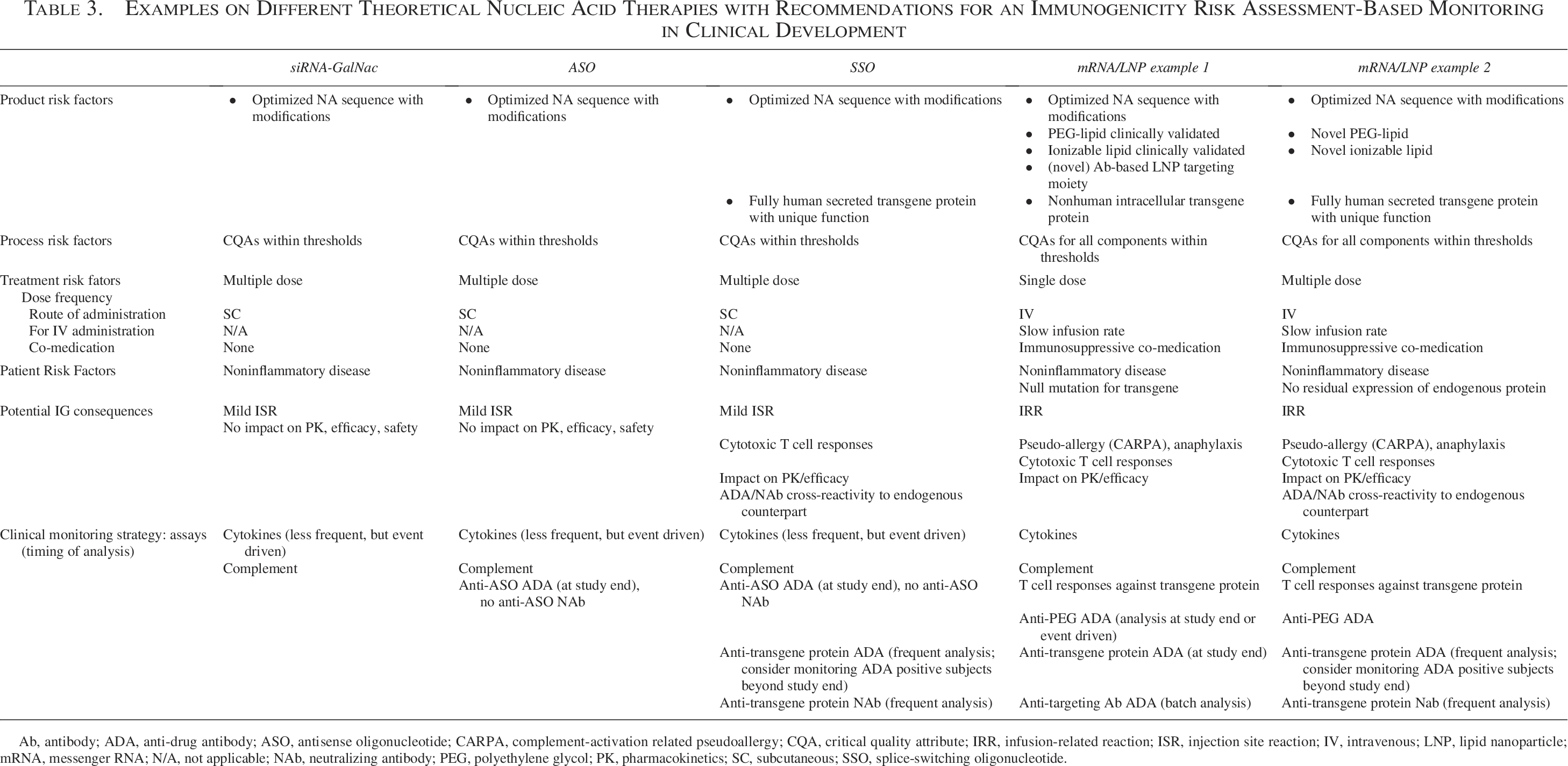
Ab, antibody; ADA, anti-drug antibody; ASO, antisense oligonucleotide; CARPA, complement-activation related pseudoallergy; CQA, critical quality attribute; IRR, infusion-related reaction; ISR, injection site reaction; IV, intravenous; LNP, lipid nanoparticle; mRNA, messenger RNA; N/A, not applicable; NAb, neutralizing antibody; PEG, polyethylene glycol; PK, pharmacokinetics; SC, subcutaneous; SSO, splice-switching oligonucleotide.
Conclusion
NA therapeutics, including ASOs, siRNAs, and mRNAs, represent a rapidly evolving field and groundbreaking advancement in precision medicine, offering novel treatment opportunities for various diseases. The IG profile of these therapies is shaped by a complex interplay of risk factors. IG outcomes for patients strongly depend on the NA modality and may have limited impact or even pose significant risks that must be carefully managed to ensure patient’s safety and benefit. Implementing the IRA early in development of NA therapies provides valuable opportunities for understanding and managing these risks. By incorporating insights from literature and existing experience, as outlined in this article, developers can design molecules with diminished IG and choose candidates with the most advantageous IG profiles. During clinical development, a tailored IRA-based IG monitoring strategy allows an adequate IG characterization and reduces the burden on patients by avoiding extensive monitoring when not needed. The continuous evolution of these strategies will be crucial in ensuring that these therapies are both safe and effective, ultimately leading to improved outcomes for patients.
Footnotes
Acknowledgments
The authors would like to thank the IQ Translational and ADME Sciences Leadership Group (IQ TALG) and the Clinical Pharmacology Leadership Group (IQ CPLG) for their support of this work. This work reflects the collective efforts of 8 member companies of the IQ Consortium Working Group on Immunogenicity of nucleic acid-based modalities. The multidisciplinary team leveraged member experience to standardize immunogenicity risk assessment for ASOs, siRNAs, and nonvaccine mRNA/LNP therapies. These efforts, which culminated in this article and in a webinar in November 2023, aim to advance the science and regulatory understanding for nucleic acid therapeutics.
Author Disclosure Statement
J.G.-G., M.B., and P.M. are employees of Bayer AG and hold shares of the company. L.-Z.C. is an employee of Boehringer Ingelheim and reports no conflict of interest. S.G. and E.T. are employees of AbbVie and hold shares of the company. S.L. is an employee of Roche Diagnostics GmbH and holds shares of the company. T.N. is an employee of Sarepta Therapeutics and reports no conflict of interest. S.T. is an employee of Pfizer Inc. and may hold shares of the company. A.S.Y. is an employee of GSK and holds shares of the company. V.J. is an employee of Bristol Myers Squibb NJ, USA and holds shares of the company.
Funding Information
Apart from the time investment, no other funding was involved in the preparation of the article.
Disclaimer
The content of this article represents the authors’ opinions and does not necessarily represent the views of their employers.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.