
Abstract
Introduction
Trasnthyretin amyloid cardiomyopathy (ATTR-CM) is an infiltrative progressive disease caused by deposition of transthyretin proteins in the interstitial space of the myocardium. The result is increased myocardial wall thickness which leads to diastolic dysfunction. Overlooked the disease and delayed diagnosis are common, especially in early onset cases. Clinical suspicious and early screening is a needed to specific scenarios to provide available treatments. ATTR-CM is one of the most common types of cardiac amyloidosis. The prevalence depends on the type of ATTR-CM; hereditary ATTR-CM (ATTRm) corresponds to a less stable mutation in TTR protein while wild-type ATTR-CM (ATTR-wt) is associated to age-related changes. ATTRm prevalence is estimated to be between <1 in 100 000 and 1 in 1 000 000. Amyloid cardiomyopathy more commonly affects men, elderly, and 3% to 4% of African-Americans.1,2 ATTRwt has been reported to be present in 25% of the population older than 85 years of age. However, some cases have been reported in patients younger than 65 years of age.1,3 We describe a 62-year-old African-American male who was screened with genetic testing for ATTRm. This helped confirm a diagnosis of ATTR-CM after being followed for several years due to arrhythmias.
Case Presentation
A 62-year-old African American male who had been an Olympic runner and a past medical history of hyperlipidemia, prostate cancer, hypertension, bilateral carpal tunnel surgery was being followed by electrophysiology due to atrial flutter and Mobitz 2 atrioventricular block. He underwent an ablation when he was 55 years old, followed by permanent peacemaker placement at age 58. Over time the patient noticed lower extremity edema, orthopnea, and dyspnea on exertion and was sent to a heart failure specialist. Transthoracic echocardiography showed a left ventricular ejection fraction of 50%, left ventricular internal diastolic dimension of 5.0 cm, left ventricular posterior wall diameter in diastole 1.2 cm, bi-atrial enlargement, with apical and septal sparing on global strain imaging. Electrocardiogram showed sinus rhythm (Figure 1), no signs of left ventricular hypertrophy. A restrictive cardiomyopathy was suspected, and medical therapy for heart failure was initiated. Due to his history of bilateral carpal tunnel, with no risk factors for this disease, and now heart failure it was decided to send screening for ATTRm. The result demonstrated a pathogenic variant in the trasnthyretin gene consistent with pV1421 (also known as C.424G>A and V122I). Non-invasive testing was sent after to corroborate myocardial infiltration. Technetium-Pyrophosphate scan was done which showed results equivocal for myocardial transthyretin amyloidosis. Per protocol serum and urine protein electrophoresis was sent out, which did not find any monoclonal proteins, and alpha globulins were within normal limits. It was then decided to proceed with a right heart catheterization and endomyocardial biopsy. Right heart catheterization showed waveforms which were suggestive of a restrictive cardiomyopathy (Figure 2), a right atrial pressure of 22 mmHg, pulmonary artery systolic pressure of 42 mmHg, pulmonary artery diastolic pressure of 25 mmHg, mean pulmonary artery pressure of 32 mmHg, pulmonary artery wedge pressure of 26 mmHg, and cardiac index of 2.1 min m2. Endomyocardial biopsies taken later revealed green birefringence of tissue with congo rain stain, consisted with amyloid deposition (Figure 3A and B). Liquid chromatography tandem mass spectrometry detected a peptide consistent with transthyretin amyloid deposition. After confirming the diagnosis aside from medical therapy to aid with heart failure symptoms he planned to receive treatment with tafamidis.
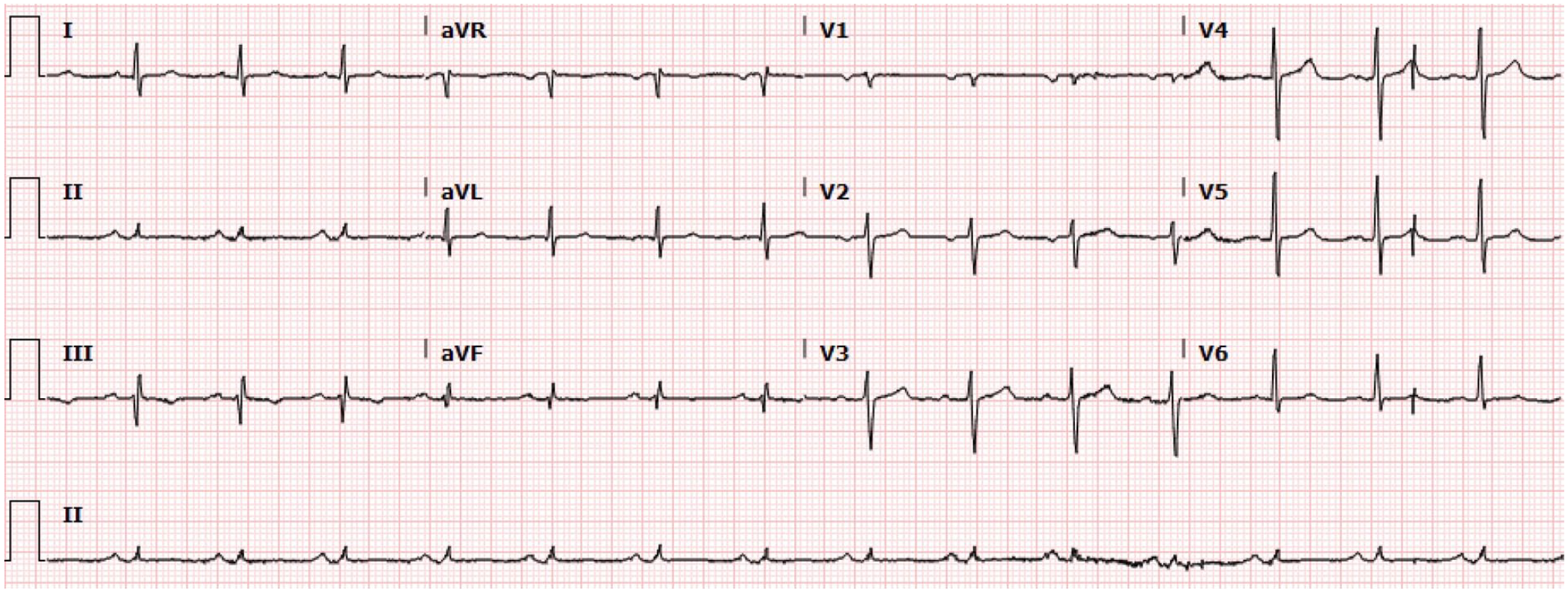
Electrocardiogram showing sinus rhythm.

Right atrial waveforms. Notice the prominent Y decent which is larger than the x decent, suggesting a restrictive filling RV defect.

Endomyocardial biopsy: (A) congo red histochemical stain shows green birefringence under cross-polarized light and (B) a sulfated Alcian blue histochemical stain demonstrated bright green staining of the vascular wall, consistent with amyloid deposition. Additional interstitial myocardial deposition is present in the upper right corner of the image.
Discussion
ATTR-CM is considered a rare disease, however its prevalence is increasing due to advances in screening tools.1,3 Regardless of screening tools advances, ATTR-CM continues to be underdiagnosed; the highest mortality rates of ATTR-CM has been seen in black men. However, lower mortality rates of ATTR-CM mortality have been seen in southern states, despite the higher proportions of black individuals in these regions. Interestingly, availability of resources, specialized physicians, and amyloidosis referral centers are key factors influencing the detection of this rare disease. In one study, which reported regional trends and geographic disparities in cardiac amyloidosis mortality, regions with amyloidosis referral center at <400 km reported the highest proportion of cardiac amyloidosis mortality cases. 4 Differences in ATTR-CM mortality has also been described in urban versus rural counties. Higher mortality rates have been identified in men living in rural counties versus urban counties. Some of the factors that might influence this trend is that rural counties had lower per capita income, higher unemployment rates, lower median household income, higher poverty rates, and higher proportion of adults 65 years or older. 5 Though, this report remarks the importance of early diagnosis of a patient with ATTR-CM in a southern region, rural county in the United States; these regions have showed lower proportions of cardiac amyloidosis but have a population with risk characteristics.4,5 ATTR-CM is more common in men, elderly and African Americans. 2 The approach to suspected cases of ATTR-CM relies on the presence of heart failure, red flag symptoms and age >65 or >70 for men and women, respectively. 1 However, younger patients than 65 or 70 years of age for men and women respectively have been detected, 6 which pose the interrogation of the importance of early genetic screening in those patients who we have a strong suspicion for this condition.
The most common symptom of ATTR-CM is heart failure; arrhythmias have also been reported as the initial manifestation in these patients. The patient described, reported arrhythmias at 55 years of age, and the development of heart failure several years later, a characteristic feature of patients with ATTR-CM reported in the literature. What mainly helped to increase the suspicion of his disease was a combination of clinical and subtle echocardiographic findings and genetic screening. ATTR-CM suspicion and diagnosis is challenging; however, awareness of the diseases is increasing, and best practices to diagnose and manage it are being proposed. Key factors to approach this pathology is the expertise of the healthcare providers on ATTR-CM, the availability of a multidisciplinary team and specialized centers.3,7
The clinical presentation of ATTR-CM cases varies according to phenotypic and genetic variability. Identifying early-onset clinical signs and symptoms is a key step in the pathway to diagnose the disease. 8 The clinical presentation of the disease and debutant signs and symptoms can be neurologic such as in V122I mutation, while in other cases it seems to start with a more cardiogenic sort of symptoms.1,8 A recent consensus of experts and other studies have recommended appropriate tests according to the suspicion signs presented at onset; some of the history and examination clues include bilateral carpal tunnel syndrome, bicep tendon rupture, unexplained peripheral neuropathy, patients >60 years of age with diastolic heart failure phenotypically similar to hypertensive heart disease or hypertrophic cardiomyopathy and unexplained atrial arrhythmias or conduction system disease or need of a pacemaker.9,10 The data demonstrating that ATTR-CM can be present in an earlier age than the expected is increasing, thus, these new different clinical scenarios would benefit from introducing the genetic screening in cases where we have a strong suspicion of ATTR-CM, to approach this rare but increasingly more common disease.
Conclusions
ATTR-CM is a rare disease with an increasing prevalence. Cases with out of proportion signs and symptoms of heart failure with diastolic dysfunction with preserved ejection fractions should raise the suspicion of ATTR-CM despite age. Lower prevalence and ATTR-CM diagnosis rates might be influence by geographic and community types.
Footnotes
Patient Consent
The patient agreed to use her case as a case report and allowed us to share her medical history, laboratory results, and images.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.