
Abstract
Introduction:
Keloids and hypertrophic scars are fibroproliferative disorders of the skin that result from abnormal healing of injured or irritated skin. Multiple studies suggest that genetic, systemic and local factors may contribute to the development and/or growth of keloids and hypertrophic scars. A key local factor may be mechanical stimuli. Here, we provide an up-to-date review of the studies on the roles that genetic variation, epigenetic modifications and mechanotransduction play in keloidogenesis.
Methods:
An English literature review was performed by searching the PubMed, Embase and Web of Science databases with the following keywords: genome-wide association study; epigenetics; non-coding RNA; microRNA; long non-coding RNA (lncRNA); DNA methylation; mechanobiology; and keloid. The searches targeted the time period between the date of database inception and July 2018.
Results:
Genetic studies identified several single-nucleotide polymorphisms and gene linkages that may contribute to keloid pathogenesis. Epigenetic modifications caused by non-coding RNAs (e.g. microRNAs and lncRNAs) and DNA methylation may also play important roles by inducing the persistent activation of keloidal fibroblasts. Mechanical forces and the ensuing cellular mechanotransduction may also influence the degree of scar formation, scar contracture and the formation/progression of keloids and hypertrophic scars.
Conclusions:
Recent research indicates that the formation/growth of keloids and hypertrophic scars associate clearly with genetic, epigenetic, systemic and local risk factors, particularly skin tension around scars. Further research into scar-related genetics, epigenetics and mechanobiology may reveal molecular, cellular or tissue-level targets that could lead to the development of more effective prophylactic and therapeutic strategies for wounds/scars in the future.
Lay Summary
Recent research indicates that the formation of keloids and hypertrophic scars clearly associates with genetic, epigenetic, systemic, and local risk factors, particularly skin tension around scars. These findings suggest that molecular, cellular, and/or tissue-level approaches that target one or more of these risk factors may be promising scar therapies. Further research into scar-related genetics, epigenetics, and mechanobiology is needed, as it is likely to help identify more effective prophylactic and clinical treatment strategies for wounds and scars.
Keywords
Introduction
Keloids and hypertrophic scars are dermal fibroproliferative disorders that are due to abnormal wound healing and are characterised by excessive deposition of collagen.1,2 These scars associate with pain, hyperaesthesia and pruritus that can dramatically affect patient quality of life, especially in the case of keloids. 3 Clinicians define keloids as scars that grow into the surrounding normal skin and hypertrophic scars as scars that do not extend beyond the boundaries of the original wound.4,5 Pathologists make a histological distinction between keloids and hypertrophic scars: thus, keloids are defined by the presence of dermal nodules plus multiple thick eosinophilic (hyalinising) collagen bundles called keloidal collagen whereas hypertrophic scars bear dermal nodules only.
Keloid and hypertrophic scar formation and progression is complex and poorly understood. However, there is now considerable evidence that keloidogenesis may be driven by multiple systemic and local factors that together or alone spur persistent inflammation in the wound and subsequent scar: this inflammation leads to chronic fibroblast activity and blocks scar maturation. The systemic factors include adolescence and pregnancy, which associate with a higher risk of bulky scar formation. 6 Pregnancy also aggravates keloids. This may reflect the vasodilatory effect of oestrogen, which may promote the movement of immune factors and cells into the wound or scar bed, thereby exacerbating local inflammation. Recent studies show that hypertension also associates with keloid aggravation.7–10 It is possible that the strain imposed by hypertension on the existing and newly forming blood vessels in keloids promotes their vasodilation and exacerbates the chronic local inflammation. 11
Local risk factors for bulky scar formation and progression include delayed wound healing, wound depth and, most importantly, mechanical forces such as the skin tension that is induced by stretching. 10 This is evidenced by the fact that keloids show a strong predisposition to occur on body areas with strong and/or repetitive stretching of the skin, namely, the anterior chest, shoulder, deltoid, jaw and ear. By contrast, keloids rarely occur in areas where the stretching of the skin is rare, such as the scalp or anterior tibiae. 12 Skin tension may also explain why keloids on the dominant sites often have characteristic growth patterns, namely, the butterfly and crab’s claw on the chest and the dumbbell on the shoulder.13–17
Keloid formation and/or progression also associate with a variety of genetic and epigenetic factors. First, a genetic predisposition is suggested by the fact that keloids are more common in dark skinned individuals and Asians. Moreover, keloid patients often have a family history of these scars. 18 Single nucleotide polymorphisms (SNPs) also associate with keloid formation.11,19–23 For example, a genome-wide association study (GWAS) 19 found that four SNP loci in three chromosomal regions associate significantly with keloids. Recent studies also suggest that a variety of epigenetic mechanisms may promote keloid formation. Some of these changes, including DNA methylation, alter the structure of the DNA and thereby shape cell division and cell phenotype.24,25 Other epigenetic changes involve non-coding RNAs that alter cell phenotype. 26
This review describes the current status of the basic research on the genetic, epigenetic and local mechanical force risk factors that drive the formation and progression of keloids and hypertrophic scars.
Methods
The PubMed, Embase and Web of Science databases were subjected to an extensive literature search using the following MESH terms: [genome-wide association study (GWAS)] and [keloid]; [single nucleotide polymorphisms (SNPs)] and [keloid]; [epigenetics] and [keloid]; [non-coding RNA] and [keloid]; [microRNA] and [keloid]; [long non-coding RNA] and [keloid]; [DNA methylation] and [keloid]; and [mechanobiology] and [keloid]. The databases were searched from their individual dates of inception through to July 2018.
Role of genetics in bulky scarring
Studies identifying genetic variants
The family pedigrees of keloid patients show that keloids may be inherited via an autosomal dominant mode with incomplete penetrance. However, it seems that keloid disease is not a simple Mendelian monogenic disease; rather, it appears to be a complex oligogenic disorder.27–30
Several studies have identified keloid-related SNPs. Thus, when Nakashima et al. conducted a GWAS in the Japanese population, they found a significant relationship between keloids and four SNPs (rs873549, rs1511412, rs940187 and rs8032158) in three chromosomal regions (1q41, 3q22.3-23 and 15q21.3). 19 Ogawa et al. then reported that one of these SNPs, namely, rs8032158, may also influence keloid severity. 20 A genome-wide linkage study found that the keloids in a Japanese family were linked to the 2q23 chromosomal region while those in an African American family were linked to 7p11. 29 However, another genome-wide linkage study on a large Chinese family with keloids did not show the linkage to 7p11; 31 by contrast, linkage intervals at 15q22.31-q23, 18q21.1 and 10q23.31 were found in this family.32,33 Notably, the 18q21.1 region harbours the mothers against decapentaplegic homologue (SMAD) 2, 7 and 4 genes, which participate in the regulation of the transforming growth factor (TGF)-β signalling pathway. Interestingly, many recent studies have reported that multiple genes in TGF signalling pathways have fibrosis-related functions: they include TGF-β1, TGF-β2, TGF-β3, TGF-β receptor (TGF-βR) I, TGF-βRII, TGF-βRIII, SMAD3, SMAD6, SMAD7, epidermal growth factor receptor (EGFR) and TNF-alpha-induced protein 6 (TNFAIP6).34–39
Several studies have shown that several HLA genes associate significantly with keloids.40–42 Of these, HLA-DRB1*15 appears to have the most robust association since a link between this HLA and keloids was observed in both Chinese and Caucasian ethnic groups.40,41 This suggests that HLA-DRB1*15 may associate with an increased risk of keloid disease. A microarray analysis and subsequent validation studies also showed that HLA-DRB5 in the 6p21.32 chromosomal region associates significantly with keloid pathogenesis. 42
In terms of other keloid-associated genes, Chung et al. showed that the NEDD4 gene in the 15q21.3 chromosomal region upregulates fibronectin and type 1 collagen expression and thereby promotes the accumulation of extracellular matrix and fibrosis. 43
It should be noted that these genetic variants only explain part of the many biological and/or functional changes that associate with keloid formation/progression. How they contribute to the molecular pathogenic mechanisms that trigger and maintain fibrosis remains unclear. For example, it is not known whether any genetic variants participate in the production of the pro-fibrotic myofibroblast phenotype, which plays a key role in keloidogenesis.
Epigenetics
There is strong evidence showing that environmental factors that shape gene expression via epigenetic mechanisms play an important role in both physiological and pathological conditions, especially tumorigenesis. 23 Recent studies also suggest that, along with genetic factors, epigenetic mechanisms may also play an important role in keloid fibrosis.44,45 These mechanisms include non-coding RNAs and DNA methylation.
Non-coding RNAs
A non-coding RNA is an RNA molecule that is not translated into a protein. Multiple types of non-coding RNAs that are abundant and regulate gene expression have been identified. They include transfer RNAs, ribosomal RNAs, small RNAs such as microRNAs (miRNAs) and silent interfering RNAs, and long non-coding RNAs (lncRNAs). 46 A recent study suggested non-coding RNAs may participate in keloid pathogenesis since keloids and keloid fibroblasts express miRNAs and lncRNAs at different levels compared to normal tissues and fibroblasts. 47
miRNAs
miRNAs are short (20–24 nucleotides), single-stranded, non-coding RNAs whose sequences are complementary to a target messenger RNA (mRNA) sequence. Consequently, when they bind to their target, they silence the gene in a post-transcriptional manner.48,49 miRNAs are thought to be dysregulated in many skin diseases, including malignant skin diseases and keloids.50–53 Several groups have subjected keloidal and normal fibroblasts to miRNA expression microarrays. One reported that nine miRNAs are expressed at different levels in keloidal fibroblasts relative to normal fibroblasts: six were upregulated (miR-152, miR-23b-3p, miR-31-5p, miR-320c, miR-30a- 5p and hsv1-miR- H7) and three were downregulated (miR-4328, miR-145-5p and miR-143-3p). 54 Significantly, Wu et al. found that miRNA-199a-5p is downregulated in keloids: they then showed that miR-199a-5p may inhibit the expression of genes involved in fibroblast proliferation by regulating the cell cycle.55,56 Moreover, Liu et al. showed that miRNA-21 is upregulated in keloid fibroblasts and may promote keloidogenesis by regulating fibroblast proliferation and apoptosis: this appears to be achieved by modulating the expression of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) via the PI3K/AKT signalling pathway.57,58
lncRNA
lncRNA are mRNA-like molecules that are > 200 nucleotides and lack functional open reading frames. lncRNAs can regulate gene expression, thereby controlling the cell cycle and cell proliferation.59,60 When Liang et al. performed a co-expression network study of lncRNA and mRNA, they found that a lncRNA called calcium voltage-gated channel subunit alpha1 G-antisense 1 (CACNA1G-AS1) was upregulated in keloids. This suggested that lncRNAs may be involved in keloid pathogenesis. 61 Moreover, Zhu et al. found that lncRNA-ATB can regulate the autocrine secretion of TGF-β2 in keloid fibroblasts by inhibiting their expression of zinc finger protein 217 (ZNF217) via miR-200c. Thus, the lncRNA-ATB/miR-200c/ZNF217/TGF-β2 signalling pathway may contain potential biomarkers and targets for novel diagnostic and therapeutic approaches to keloid disease. 62 Similarly, Sun et al. recently identified four skin-related lncRNAs (CACNA1G-AS1, LINC00312, HOXA11-AS and RP11-91I11.1) that are involved in Wnt-gene regulation in keloid disease. They proposed that the excessive proliferation in keloids may be due to lncRNA-mediated regulation of Wnt signalling pathways. 63
DNA methylation
DNA methylation is the most common form of epigenetic modification. Increased methylation (hypermethylation) turns off genes while decreased methylation (hypomethylation) turns them on. The methylation process is catalysed by DNA methyltransferase (DNMT) enzymes.64,65 The most common site of methylation is at CpG dinucleotides. 66 In mammalian cells, there are three DNMT isoforms: DNMT1; DNMT3a; and DNMT3b.
Many recent studies have shown that DNA methylation may affect fibroblast differentiation and tissue fibrosis.67–69 In particular, Hu et al. reported that DNMT-mediated DNA methylation regulates α-SMA gene expression during myofibroblast differentiation. 70 5-aza-2-deoxycytidine (5-aza-dC) is an inhibitor of DNMT. Fu et al. investigated fibrosis correlated with DNA hypomethylation which may result from upregulation of TGF-β type I receptor. These results suggest that regulating DNA methylation is important in initiation of scar formation.71,72 A similar relationship between DNA hypermethylation and elevated fibrotic signalling was found by Yang et al.: not only did 100% of keloid fibroblasts express DNMT1 versus 8% of normal skin fibroblasts, keloid lesions also expressed higher levels of the fibrotic genes TGF-β1, phospho-smad2 and phospho-smad3 and lower levels of the anti-fibrotic gene smad7. Notably, 5-aza-2-deoxycytidine treatment inverted the expression patterns of DNMT1, TGF-β1 and smad7. 73 Moreover, when Jones et al. subjected six keloid and six normal skin samples to genome-wide profiling with the Infinium HumanMethylation450 BeadChip platform, they found that the keloid genomes were overall more likely to be hypomethylated rather than hypermethylated in non-promoter genomic regions. In addition, of the 197 CpGs that were differentially methylated in keloids, 191 were in the Ingenuity Pathway Analysis (IPA) database and mapped to 152 unique genes.74,75 Causal Network Analysis software then showed that four master regulators (pyroxamide, tributyrin, PRKG2 and PENK) and 19 intermediate regulators may play key roles in keloid pathogenesis. 76
Thus, DNA methylation and other epigenetic mechanisms may play an important role in the pathogenesis of keloid disease. This may also explain why gene mutation analyses alone have not provided any definitive mechanisms that underlie keloid formation.74–76
Mechanobiology
Role of mechanobiology in cutaneous wound healing and scarring
The cutaneous wound-healing process consists of the inflammatory, proliferative and remodelling phases. While the mechanisms that regulate wound healing remain to be fully understood, 77 it is now clear that mechanical forces play important roles in normal wound healing. For example, myofibroblasts exert an appropriate amount of intrinsic tension that causes the wound to contract and close. Moreover, normal mechanical forces on and in the wound/scar induce a homeostatic state of ‘tensegrity’ (tensional integrity) that allows the cells and the extracellular matrix in the tissue to progress normally through the various phases of wound healing. However, when large or unusual extrinsic mechanical forces (e.g. scratching, compression and strong/repetitive skin stretching) are placed on the healing wound or scar, the tensegrity becomes dysregulated and bulky scarring can ensue.78–80
Role of mechanobiology in the formation and progression of keloids and hypertrophic scars
Recent research suggests that hypertrophic scars and keloids are caused by the same fibroproliferative pathology and that their different clinical and pathological features largely reflect the degree of inflammation in the healing wound/scar. This inflammation is in turn determined by various risk factors, including systemic and local mechanical forces (and possibly also genetic and epigenetic factors). This hypothesis suggests that mature scars, hypertrophic scars and keloids can convert into each other during the clinical course and vice versa. Indeed, keloids can grow from mature scars and we have encountered many cases of bulky scars that bear clinical and histological features of both keloids and hypertrophic scars (personal observations).
There is now substantial evidence that a key risk factor for both the formation and progression of bulky scars is mechanical force. First, hypertrophic scars can be generated in experimental animal models by repeatedly applying mechanical forces on the edge of an incisional wound. 4 Second, a statistical study of 1500 anatomic regions in Asian patients showed that keloids tend to occur on specific body sites (Figure 1), namely, the anterior chest, shoulder, scapula and lower abdomen–suprapubic region. 17 All of these body sites are characterised by strong and/or repetitive stretching of the skin. Thus, the anterior chest skin is regularly stretched horizontally by the pectoralis major muscle, the shoulder and scapula skin is repeatedly stretched by upper-limb movements and bending of the body, and the lower abdomen-suprapubic skin is stretched many times a day due to changes in position (e.g. sitting and standing). By contrast, body sites that experience little skin tension (e.g. the scalp, upper eyelid and anterior lower leg) rarely bear keloids. Third, keloids tend to spread into distinctive shapes that depend on their location. For example, keloids on the anterior chest tend to form a crab’s claw shape, shoulder keloids develop butterfly shapes and upper arm keloids often grow into a dumbbell shape. These patterns may reflect the different predominant directions of skin tension at these sites. 13 Indeed, our finite-element analysis of the distribution of the mechanical forces around a scapular keloid showed that the keloid had expanded in the direction of dominant skin-pulling. Moreover, the stiffness of the skin at various points at the keloid circumference correlated directly with the degree of skin tension at these points.
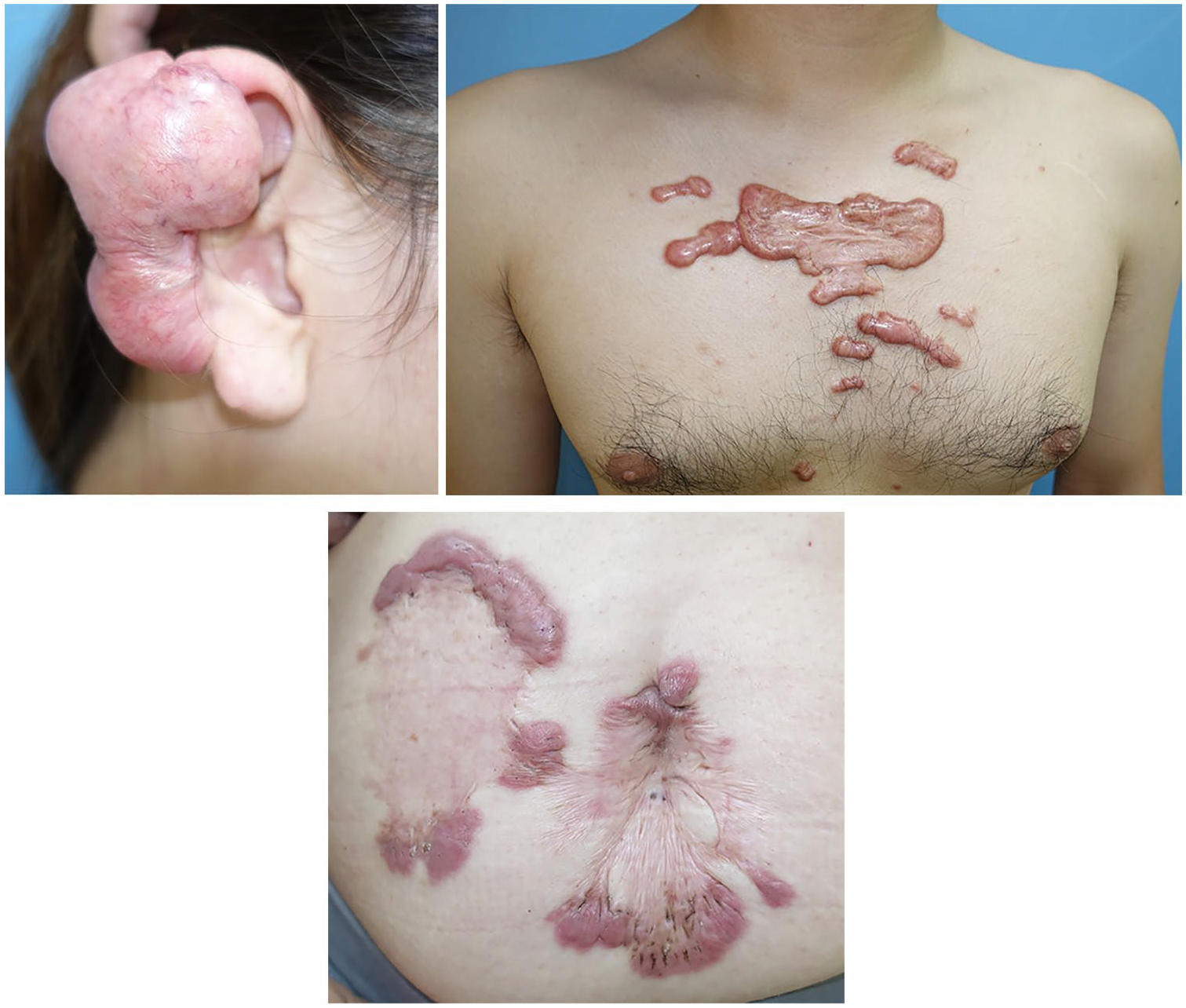
Typical keloids. Keloids occur at specific sites including the ear, anterior chest wall, shoulder and lower abdomen. The direction of keloid growth results in characteristic shapes, which depend on their location. For example, keloids on the anterior chest grow horizontally in a ‘crab’s claw’-like pattern and this horizontal direction of tension is caused by pectoralis major muscle contraction. Keloids on the lower abdomen grow vertical and this direction is caused by rectus abdominis muscle movement.
The relationship between mechanical force on the wound/scar and local inflammation is clearly depicted by combining the finite-element analysis data mentioned above with our histological examinations of keloids.13,16 The finite-element analysis showed that there is high skin tension at the keloid edges and lower tension at the centre, while our histological analyses indicated that the key features of keloid inflammation (e.g. angiogenesis, fibroblasts and inflammatory cells) gradually waned in intensity as the histological keloid sections moved from the periphery to the centre. Thus, it seems that skin tension dictates the degree of inflammation, which in turn determines the pattern and degree of keloid growth.
Mechanosignalling pathways in cutaneous scarring
Analyses of fibroblasts harvested from human or animal scar tissues have revealed a variety of mechanosignalling pathways that appear to participate in the formation and growth of cutaneous scars. The TGF-β/Smad, integrin and calcium ion pathways are now well-known to participate in scarring. Other pathways that may play a role include the MAPK and G-protein, Wnt/β-catenin, TNF-α/NF-κB and IL pathways: however, further research on these pathways is needed. Various lines of evidence suggest that during scar formation, these cellular mechanosignalling pathways interact actively with the extracellular matrix and crosstalk extensively with hypoxia, inflammation and angiogenesis pathways. Wong et al. showed that other participating signalling pathways may be the focal adhesion kinase (FAK)-extracellular-related kinase-monocyte chemoattractant protein-1 pathways: they showed that these pathways are activated by mechanical forces and mediate fibrosis in the healing wound. 78 Further elucidation of the mechanosignalling pathways that contribute to normal and abnormal scarring will help us to better understand scar development. This in turn may facilitate research into this promising field and may aid the development of pharmacological interventions that can limit, minimise or even reverse scar formation and progression.81–84
Conclusion
Recent research indicates that the formation of keloids and hypertrophic scars clearly associates with genetic, epigenetic, systemic and local risk factors, particularly skin tension around scars. These findings suggest that molecular, cellular and/or tissue-level approaches that target one or more of these risk factors may be promising scar therapies. Indeed, there is increasing evidence that these approaches may be effective in the clinic. Despite this, the current clinical treatment strategies continue to focus on decreasing inflammatory processes. Further research into scar-related genetics, epigenetics and mechanobiology is needed, as it is likely to help identify more effective prophylactic and clinical treatment strategies for wounds and scars.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.