
Abstract
Background:
Burn injury represents a significant public health problem worldwide. More than in any other injury, the inflammation and catabolism associated with severe burns can exacerbate nutrient deficiencies resulting in impaired immune function and increased risk of developing infection, organ dysfunction and death. Consequently, over the last few decades numerous trials have evaluated the impact of different nutritional strategies in severe burn injury. Glutamine is of particular interest, as it appears vital for a number of key stress-response pathways in serious illness. The purpose of the current manuscript is to provide the rationale and protocol for a large clinical trial of supplemental enteral glutamine in 2700 severe burn-injured patients.
Methods:
We propose a multicentre, double-blind, pragmatic, randomized, clinical trial involving 80 tertiary intensive care unit (ICU) burn centres worldwide. We aim to enrol patients with deep second- and/or third-degree burns at moderate or high risk for death. We will exclude patients admitted > 72 h before screening and patients with advanced liver and kidney disease. The study intervention consists of enteral glutamine 0.5 g/kg/day vs. isocaloric maltodextran control delivered enterally. Primary outcome will be six-month mortality. Key secondary outcomes include time to discharge alive from hospital, ICU and hospital mortality, length of stay and health-related quality of life at six months.
Significance:
This study will be the first large international multicentre trial examining the effects of glutamine in burn patients. Negative or positive, the results of this trial will inform the clinical practice of burns care worldwide.
Clinicaltrials.gov ID #NCT00985205
Lay Summary
Patients with severe burns need to recover in a hospital burn unit for a long time and are at high risk of developing infections and dying. Proper nutrition and certain nutrients may improve survival in these patients and shorten their stay in the burn unit. Glutamine is a building block of protein that is normally made in the body and is found in different foods we eat. It is of great interest because it has several beneficial effects on the body during serious illness, such as with burn injury. In this study, we will look at the effect of glutamine supplementation on survival and time spent in hospital. A total of 80 burn units around the world will enrol 2700 patients with 2nd or 3rd degree burns over 4 years. Patients will receive either glutamine powder or a placebo through a feeding tube or mixed with food, from admission to the burn unit, until a week after the burn wound has healed. The main outcome for this study is survival at 6 months. Other outcomes include the time taken to be discharged from hospital alive and duration of stay in the burn unit. This study will be the first large international multicentre trial examining the effects of glutamine in burn patients. Glutamine may lead to better survival and less complications in burn patients, who have a devastating and disabling burn injury. If the trial is positive, the results will be used to inform how nutrition should be given to such patients worldwide.
Introduction
Worldwide, burn injuries represent a significant public health problem and are ranked the fourth most common injury. 1 Burn injury strikes largely young to middle-aged working people, resulting in the leading cause of disability adjusted life years in low and middle-income countries.1,2 The incidence of hospital visits caused by burn injuries was 220 per 100,000 habitants during 1993–2004. 3 Since the mid-1980s, mortality from burn injuries has plateaued and the leading cause of death from burn injuries continues to be multiple organ failure and sepsis.4,5 In severe burns and other critical illnesses, the relationship between nutrient deficiencies, altered immune status and acquired infection has been recognised for many years. More than in any other injury, the inflammation and catabolism associated with severe burns can exacerbate nutrient deficiencies, thereby predisposing patients to impaired immune function and increased risk of developing infectious complications, organ dysfunction and death. Consequently, over the last few decades numerous trials have evaluated the impact of different nutrition/nutrient strategies in critically ill patients and in particular, severely burn-injured patients.6,7 Glutamine is of particular interest in this regard as it appears vital for a number of key stress-response pathways in serious illness. 8 Observational studies have shown that glutamine levels decrease acutely during critical illness 9 and that low levels of glutamine are associated with immune dysfunction 10 and increased mortality.11,12 In a single-centre pilot trial of enteral glutamine, the enteral glutamine supplementation showed a threefold reduction in positive blood cultures and a dramatic reduction in mortality in severe burn patients. 13 Six other randomized trials of glutamine supplementation in burns patients have been conducted, and when summarised, they are suggestive of a significant reduction in mortality and hospital length of stay. 14 With this limited evidence, many practitioners are prescribing enteral glutamine to burn-injured patients. 15 However, as explained below, randomized controlled trials in other critically ill patient populations have suggested that glutamine administration may be harmful and increase mortality.16,17 Given this conflicting evidence, burn practitioners are either harming or saving lives with glutamine use. It is imperative that we determine the efficacy and safety of glutamine now!
The purpose of the current manuscript is to provide the rationale for and methodological details of a large, multicentre clinical trial of supplemental enteral glutamine in 2700 severe burn-injury patients. We hypothesise that the inexpensive therapeutic strategy tested in this randomized controlled trial will lead to lower morbidity and mortality and reduced healthcare costs in an otherwise very devastating and disabling injury worldwide. This trial will be the largest trial of burn-injured patients ever conducted.
Background rationale and systematic review of the literature
The amino acid glutamine plays a central role in nitrogen transport within the body, is a fuel for rapidly dividing cells (particularly lymphocytes and enterocytes), is a precursor to glutathione and nucleotides, and has many other essential metabolic functions. 18 A summary of potential mechanisms of benefit for glutamine in burn injury is shown in Figure 1.18,19 Under normal physiological conditions, glutamine is synthesised in sufficient amounts by the skeletal muscle and therefore is considered non-essential. It had been hypothesised that glutamine may become a conditionally essential amino acid in patients with catabolic disease as studies indicate glutamine levels fall following major surgery,20,21 critical illness 22 and burn injury.23,24 Lower levels of glutamine have been associated with immune dysfunction 11 and increased mortality. 12

Mechanisms of glutamine’s potential benefit after thermal burn injury.
Following burn injury, recent experimental data show that glutamine supplementation reduces lymphocyte apoptosis, improves immune function and improves post-burn survival. 25 Regardless of the mechanism, many animal studies have demonstrated improved survival associated with glutamine supplementation in models of sepsis and injury.26–29 Experimental data have further shown that glutamine can reduce long-term lung injury from burn-related smoke inhalation 30 and can reduce cardiac injury and improve cardiac function following severe burn injury. 31 In addition, randomized clinical trials have shown that glutamine administration improves insulin sensitivity and protein metabolism in critically ill and in trauma patients.32–34 Finally, glutamine supplementation, when combined with post-intensive care unit (ICU) physiotherapy and adequate protein delivery, has recently been shown to improve 6-min walk test times vs. physiotherapy alone. 29 These findings suggest that glutamine may help to preserve muscle mass and long-term physical function in burn patients.
Over the past few years, we have conducted several systematic reviews and meta-analyses of existing studies of glutamine supplementation in critically ill patients.15,35–37 When the results from all randomized trials providing intravenous glutamine to all critically ill patients were aggregated, we observed a trend towards a reduction in mortality (risk ratio [RR] = 0.87, 95% confidence interval [CI] = 0.75–1.01, P = 0.07, 27 trials), a significant reduction in hospital mortality (RR = 0.70, 95% CI = 0.53–0.92, P = 0.01, 15 trials), a trend towards a reduction in infections (RR = 0.89, 95% CI = 0.77–1.03, P = 0.12, 13 trials) and a significant reduction in hospital length of stay (weighted mean difference in days [WMD] = –2.56, 95% CI = –4.71– –0.42, P = 0.02, 11 trials) associated with glutamine. 38 The analysis of the six trials in 225 burn patients shows a significant effect on mortality (RR = 0.22, 95% CI = 0.07–0.62, P = 0.005, four trials), no effect on infections (RR = 0.78, 95% CI = 0.46–1.31, P = 0.34, three trials) but a significant reduction in hospital length of stay (WMD = 6.06, 95% CI = –9.91– –2.20, P = 0.002, five trials). 15
In summary, the biologic rationale and clinical trial data we have systematically reviewed shows plausible mechanistic hypotheses and potential clinically significant benefits, thus clearly justify moving forward with this large randomized controlled trial. Taken together, there is a signal from these trials for enteral glutamine and specifically, in the burn population, glutamine supplementation may result in improved clinical outcomes. However, this signal requires confirmation in a large, high-quality, multicentre trial because of the potential for harm.
The REDOXS study: the first signal of harm
As summarised above, there is no signal to date of glutamine causing harm in burn-injured patients. However, some of our team have recently published the results of the REDOXS trial that suggested glutamine may be harmful in patients with multi-organ failure. 16 In a blinded 2 × 2 factorial trial involving 40 ICUs in Canada, the United States and Europe, we randomized 1223 critically ill, mechanically ventilated adult patients with multi-organ failure to glutamine supplementation or no glutamine and antioxidants or no antioxidants. High doses were used for glutamine: 0.35 g/kg/day of glutamine intravenously based on ideal body weight, provided as 0.50 g/kg/day of the dipeptide alanyl-glutamine (Dipeptiven®, Fresenius Kabi) and an additional 30 g/day delivered enterally, provided as alanyl-glutamine and glycine-glutamine dipeptides or respective placebo. In addition, patients were randomized to receive 500 µg of selenium intravenously (selenase®, biosyn), and the following vitamins and minerals administered enterally: selenium 300 µg, zinc 20 mg, beta carotene 10 mg, vitamin E 500 mg and vitamin C 1500 mg, or placebo. In contrast to our hypotheses and expectations, we demonstrated increased harm associated with glutamine supplementation. In addition, in a second manuscript, we conducted several post hoc subgroup analyses to identify any potentially important subgroup effects. 39 It appears that harm from both glutamine and AOX supplementation were limited to patients with renal dysfunction upon study enrolment. This harmful effect seemingly was partially ameliorated by the presence of dialysis.
The Metaplus study: another signal of harm
More recently, another trial of glutamine supplementation suggests increased mortality (Metaplus study 17 ). Van Zanten et al. recently completed a randomized trial of 300 patients comparing an enteral feeding product enriched with glutamine, omega-3 fatty acids and antioxidants compared to a high protein enteral diet. No statistically significant differences were observed in infections (primary endpoint) or other endpoints except that patients that received the enriched diet had increased higher six-month mortality rate (adjusted hazard ratio [HR] = 1.57, 95% CI = 1.03–2.39; P = 0.04), In addition, in a pre-specified sub-group analysis, six-month mortality was higher in medical patients receiving the glutamine supplemented formula compared to controls (53.7% vs. 34.5%, P = 0.044).
Differences between REDOXS and Metaplus trials and the RE-ENERGIZE trial
It is key to stress that burn-injured patients are quite different metabolically, and from a protein/amino acid loss standpoint, vs. the REDOXS and Metaplus study patients. The REDOXS trial enrolled only mechanically ventilated patients with critical illness admitted to the ICU with two or more organ failures related to their acute illness. Major burn patients were excluded from the REDOXS trial because it was considered at the time to be unethical to withhold glutamine in this patient population. This led to a population that was 80% medical ICU patients in REDOXS, while the Metaplus study only showed harm in medical ICU patients and not in surgical patients. In addition, in the REDOXS trial, 35% of the group presented in renal failure and nearly 95% of patients were on vasopressors at enrolment. The average age of the patient was ~ 63 years. This is quite different from the proposed RE-ENERGIZE trial where burns patients will be, for the most part, young and healthy before admission. The average age of patients in the pilot study so far is 48 years. The dose of glutamine used in the REDOXS trial was the highest dose ever given in a major clinical trial and was provided both enterally and parenterally. The RE-ENERGIZE trial gives 0.5 g/kg/d of L-GLN enterally (only) in the glutamine treatment arm. Enterally administered glutamine has a much lower systemic uptake than parenteral glutamine as the majority of the glutamine (~ 80%) is metabolised by the GI tract and the liver before reaching the systemic circulation. This is quite different from the systemic effects of intravenous glutamine.
Another key difference between REDOXS and RE-ENERGIZE is the baseline plasma glutamine levels. In a sub-study of 68 patients in the REDOXS trial with plasma glutamine measurements, only 31% of patients presented with low baseline glutamine levels (normal values 420–700 µmol/L). In fact, we observed supra-normal levels (> 930 µmol/L) of plasma glutamine at baseline in 15% of patients, a phenomenon only recently described and also shown to be associated with increased mortality. 40 We have not seen high plasma levels in burn-injury patients. Historically, burn patients have significant glutamine deficiency early in their stay due to massive glutamine loss via the burn wound and severe catabolism.14,32 In the context of our RE-ENERGIZE pilot study, we drew blood on 18 randomly selected patients to measure baseline plasma glutamine levels. The average level was below the lower range of normal at 408 ± 146 umol/L (normal values 420–700 µmol/L), indicating severe glutamine deficiency in our pilot burn patients, which was quite different from the REDOXS trial where very little baseline deficiency was observed. In our pilot trial of severe burn injury, the highest baseline glutamine level was 723 umol/L (within normal range).
In summary, recent studies provide us with the first signal that glutamine may be harmful in some patient populations. This signal is only apparent in large scale, multicentre trials and in non-burn populations who have normal to high levels of serum glutamine. Juxtaposed next to the systematic review of enteral glutamine in burns, which suggested reduced mortality and infections and shorter LOS, there is considerable uncertainty about the safety and efficacy of glutamine in this burn patient population. While there is no signal of harm to date in the burn population, the signal of benefit comes from six small, single-centre RCTs evaluating a total of 225 burn patients but only three trials reporting any deaths in either group. All of these trials had less than 50 patients each and were grossly underpowered to rule out harm. When statistically aggregated, these few trials do suggest a very large and implausible treatment effect. We consider this estimate of treatment effect unstable and possibly biased. Given the uncertain effect of glutamine in burn patients, including both a potential for benefit and harm, we need to conduct a large scale, multicentre trial of glutamine in burns to resolve the uncertainty about the overall effect.
The proposed methods
We propose a large, international, multicentre, double-blind, pragmatic, randomized controlled trial of 2700 patients with severe burns randomly allocated to receive enteral glutamine or placebo. We aim to recruit patients from burn or ICUs from across North America, South America, Europe and Asia. Given the large sample size across numerous participating units, we have adopted a pragmatic philosophy in developing and executing this trial protocol. Consistent with this philosophy, we have made efforts to standardise nutrition practices across all site but make no effort to standard other key aspects of burn care across these multiple burn units.
What are the planned trial interventions?
Patients will be allocated to two groups:
the glutamine group: patients will receive glutamine through their feeding tube, every 4 h or three times a day if taking things by mouth, for a total of 0.5 g/kg/day for patients with a body mass index (BMI) < 35 kg/m2. Patients with a BMI ⩾ 35 will receive 0.5 g/kg/day based on an obesity-adjusted body weight. The glutamine powder will be supplied in pre-packaged aliquots and will be delivered to the ICU in blinded sachets and will be mixed in water or other liquids at the bedside by the patient’s nurse;
the control group: patients will receive an iso-calorically delivered amount of maltodextrin (control) mixed with water or other liquids. Maltodextrin is a source of carbohydrate commonly found in standard enteral nutrition and has no metabolic effects other than serving as a trivial source of additional energy. Moreover, maltodextrin appearance makes it indistinguishable from glutamine, allowing for a double-blind design. The enteral route of glutamine administration seems preferable because our result on mortality was obtained in the pilot study using this mode of administration. 13 Glutamine or control supplement will be given within 2 h of randomisation (where possible) through enteral route (feeding tube or orally). Administration of the glutamine may be interrupted by surgical procedures. A missed administration will be compensated for by a double dose at the next dosing or a make-up dose during a later part of the day. Glutamine will not be counted in the protein intake of the patients, so the dietitian can remain blinded and prescribe protein as per usual standards of care. The justifications for not giving an isonitrogenous placebo is based on the fact that patients from both groups will receive an adequate amount of proteins through standard nutritional care and non-essential amino acids used in such a placebo may have active metabolic and cellular effects.
What are the proposed arrangements for allocating participants to trial groups?
Informed consent will be obtained within 72 h following admission to the local ICU or burn unit from the patient or substitute decision-maker in accordance with local ethics committee regulations. Once consent is obtained and necessary baseline data collected, the study coordinator will log on to the web-based randomisation system at the Clinical Evaluation Research Unit (http://www.ceru.ca/) at Kingston General Hospital. The system will confirm eligibility before allowing randomisation. The system will then provide the study coordinator with a patient study number and will send the local pharmacist notification of randomisation and treatment assignment. Allocation will be random and concealed and will be blinded to everyone except the pharmacist at each site who will be responsible for preparing study samples and delivering them to the ICU in a blinded fashion in accordance with the documented study operating procedures. The randomisation system will use a computer-generated randomisation schedule allocating patients 1:1 to either glutamine or matching placebo by the method of permuted blocks of random undisclosed size within strata. Randomisation will be stratified by site to ensure balance of treatment assignments within each site. Given the large pragmatic nature of the trial, we will not stratify by additional factors such as burn severity. Because of the large sample size, such additional stratification variables will not be helpful in balancing out the two groups, adds statistical complexity to the analysis, and add operational challenges to the conduct of the trial as all the stratification variables need to be clearly and accurately defined before randomisation.
What are the proposed methods for protecting against other sources of bias?
All research and clinical personnel at the study site with the exception of the site pharmacist will be blinded to treatment allocations. Given the nature of our study intervention, it is important that we standardise the practice of nutrition therapy in participating units. As per standard of care in these burn units, these patients will be fed enterally and for those fed into the stomach, they will have routine evaluation of gastric residual volume during feeding. To improve compliance, national nutrition guidelines, pre-printed orders and bedside algorithms are provided. Also, the use of enteral solutions that contain glutamine is restricted in all enrolled participants. Although other aspects of burn care will not be standardised, we will capture key process of care issues in our data collection strategies.
What are the planned inclusion/exclusion criteria?
Inclusion criteria consist of: deep second- and/or third-degree burns requiring skin grafting. For patients aged 18–39 years, we require a TBSA (total burn surface area) ⩾ 20% or a minimum of 15% TBSA when concomitant inhalation injury is present. For patients aged 40–59 years, we require a TBSA ⩾ 15%. For patients aged 60 years or older, we require a TBSA ⩾ 10%. Outside these limits we believe that the risk of death is too small, increasing the risk of beta error.
Exclusion criteria consist of:
72 h from admission to ICU or burn unit to time of consent;
patients aged less than 18 years;
in patients without known renal disease, renal dysfunction defined as a serum creatinine > 171 mmol/L or a urine output < 500 mL in the last 24 h (or 80 mL in the last 4 h if a 24-h period of observation is not available). In patients with acute on chronic renal failure (pre-dialysis), an absolute increase of > 80 mmol/L from baseline or pre-admission creatinine or a urine output of < 500 mL in the last 24 h (or 80 mL in last 4 h) will be required. Patients with chronic renal failure on dialysis will be excluded;
liver cirrhosis - Child’s class C liver disease;
pregnancy (urine/blood tests for pregnancy will be done on all women of childbearing age by each site as part of standard ICU practice);
contra-indication for EN: intestinal occlusion or perforation, intra-abdominal injury;
patients with injuries from high voltage electrical contact;
patients who are moribund (not expected to survive the next 72 h);
patients with extreme body sizes: BMI < 18 or > 50;
enrolment in another industry sponsored ICU intervention study (co-enrolment in academic studies will be considered on a case-by-case basis);
received glutamine supplement for > 24 h before randomisation;
known allergy to maltodextrin, corn starch, corn, corn products or glutamine.
These criteria are designed to include those severe burn-injury patients who are likely to benefit from the therapeutic intervention tested in this trial. Patients who develop renal failure during study treatment will be evaluated daily to determine if the study medication should continue. If dialysis is planned for that day or the next, study medication will continue. If not, and the clinical team is concerned about a high urea, then the study medication will be suspended for 1–2 days until dialysis occurs or the renal failure resolves.
What is the proposed duration of treatment period?
Since our initial single-centre pilot trial, we have been supplementing with glutamine until all burn wounds are excised and covered. The rationale for this choice is based on the observed effects of glutamine on infection in published studies on burn patients. Since severe infectious episodes could occur throughout the healing period we choose to give glutamine as long as the risk for infection is present. Without objective and standardised methods to measure wound-healing time, we choose to operationalise this concept by administering the study intervention to the patients until seven days after the last grafting operation or until discharge from the acute care unit or three months from admission, whichever comes first.
What is the proposed duration of follow-up?
Patient clinical status will be monitored daily during the acute care unit stay. Once discharged from the burn unit or ICU, patients will no longer be followed daily but hospital outcomes, as explained below, will be abstracted from the chart. Since glutamine may have long-term positive effects, 41 and the resolution of the burn itself, in terms of recovery from injury, may take months, we will follow patients for up to six months documenting survival and health-related quality of life (HRQOL). Diagram of study duration and follow-up are presented in Figure 2.
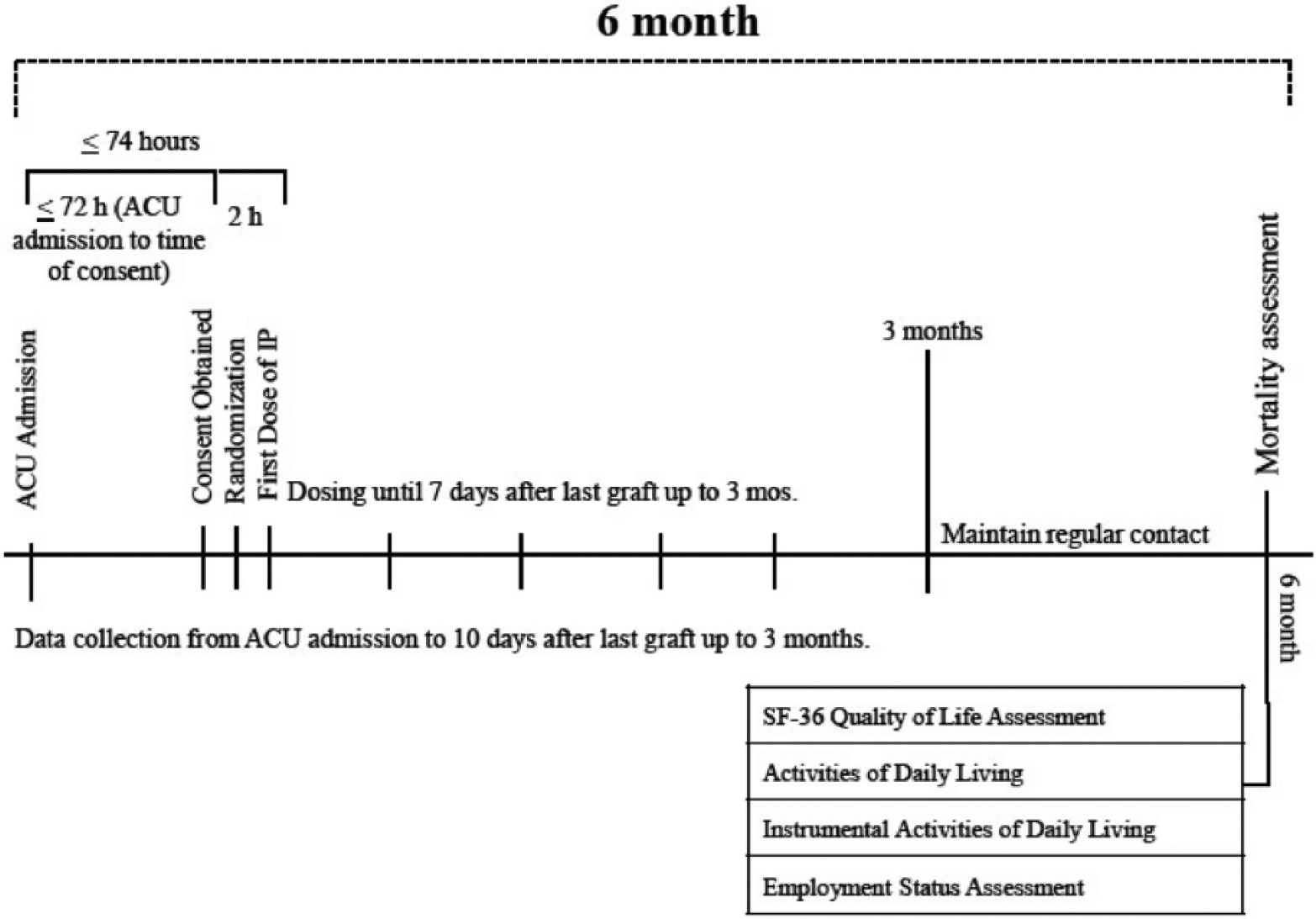
Flow diagram of study duration, since hospital admission of participants until six months of follow-up.
What are the proposed primary and secondary outcome measures?
The primary outcome for this trial is six-month mortality. We justify this endpoint as the primary outcome because of the following reasons: (1) mortality rates are still significant, particularly in low-income countries; (2) if we postulate that enteral glutamine may be harmful based on the results of recent trials, we must be adequately powered to detect such a treatment effect; and (3) it is a more objective endpoint than infection or length of stay (which are also influenced by non-clinical factors). We justify an outcome assessment at six months, as opposed to shorter time points, because a significant proportion of patients will stay more than 90 days in hospital and glutamine has been shown to have effects on six-month mortality. 41
The secondary outcome is time to discharge alive (TTDA) from hospital. TTDA is an important secondary outcome that is a composite of mortality and length of stay. This composite is similar to ‘ventilator- free days’, which is a widely accepted and commonly used outcome in intensive care research.42,43 As per the stated hypotheses of the trial, we expect glutamine to reduce infection, reduce mortality and shorten length of stay. Infections may both negatively impact length of stay and mortality, but this will be captured in a TTDA endpoint. Tertiary outcomes include HRQOL and, in particular, the physical function domain of the Short Form-36 (SF-36) questionnaire, activities of daily living (ADL) and instrumental ADL, incidence of acquired bacteraemia due to Gram-negative organisms, hospital mortality and duration of mechanical ventilation, ICU stay and hospital stay. We will also record frequency of operative procedures for burn care and other major cost drivers as outlined in Appendix 1 to support our economic evaluation.
All hospital outcomes (i.e. mortality, length of care and the incidence of bacteraemia) will be collected until discharge to home or alternative level of care (i.e. rehabilitation facility) or three months from burn unit admission, whatever comes first. To better understand the impact of the study treatment on longer-term survival, function and QOL, we will follow surviving study patients for six months. At six months after randomisation, a trained research coordinator at each site will contact patients discharged from hospital to assess survival status, whether they have resumed normal activities and administer the SF-36 over the phone. 44 To assess whether they have resumed activities, we will ask about if employed patients have returned to work and ask all patients about ADL using the Katz Index 45 and instrumental ADL using the Lawton Index. 46 We expect minimal loss to follow-up (LTFU) because many of these patients will be seen post discharge by the multidisciplinary burn team. We will further minimise LTFU by obtaining contact information for family members, in addition to the patient, at time of consent. Finally, if we cannot reach patient nor family, we will use hospital records and existing registries to ascertain patient status.
What is the proposed sample size and what is the justification for the assumptions underlying the power calculation?
Data support our belief that the mortality rate in the placebo arm will be at least 15%. This is a conservative estimate as the control group mortality rate in the meta-analysis of enteral glutamine trials in burn injury is 25% (Appendix 2). Furthermore, considering all patients with a burn injury > 20% TBSA in the American Burn Association registry, (similar to our inclusion criteria), the mortality rate is approximately 27%. We will conservatively assume a control arm mortality rate of 15% to allow for the likelihood that our mortality rate is lower than previously reported in similar patients. We consider a 25% relative risk (RR) reduction from 15% to 11.25% to be minimally clinically important. The results of the meta-analysis of existing glutamine trials in burn injury (Appendix 2) suggest that an even larger treatment effect is plausible. Smaller differences may be clinical important but may be impractical to detect and may not be large enough to shift existing practice patterns. In order to achieve 80% power to detect such a difference at two-sided alpha = 0.05 using a Chi-squared test (or two independent proportion z-test), we would need 1273 patients per arm. We will enrol a total of 2700 patients to conservatively allow for 5% LTFU.
A significant mortality effect would trump the secondary outcome of TTDA. However, based on a simulation study, we estimated that if there was no mortality difference between arms, the proposed sample size would achieve 80% power to detect a difference in TTDA if the daily hazard rate of discharge among survivors was multiplied by 1.16 for EN glutamine patients.
What is the proposed type of analyses?
The primary analysis of six-month mortality will be compared between arms using the z-test for two independent proportions. This is equivalent to the Chi-squared test for symmetric two-sided tests, but will allow us to implement one-sided interim analyses to test for increased mortality in the glutamine arm. A secondary analysis will employ the generalised mixed effects model with a random site effect. This will provide a within site interpretation of effect, will allow us to explore between site heterogeneity and will meet regulatory guidance suggesting that site be incorporated in a sensitivity analysis if it is not used for the primary analysis.47–49
The secondary outcome of this study is time to live discharge from hospital. Patients who die before hospital discharge will be treated as never being discharged from hospital by censoring them at day 181 (after last follow-up). The primary analysis will use the log-rank test. Since the log-rank test is rank based, the actual time value we assign to decedents is unimportant; they are simply considered worse (higher rank) than any patients discharged by six months. We expect minimal LTFU before hospital discharge, but if LTFU does occur due to hospital transfer or other reasons, patients will be censored at the last time known to be in the hospital. A secondary analysis will use a shared frailty model to incorporate site as a random effect. 50 The methods used for the primary (excluding the interim analyses) and secondary outcome will be applied to the binary and time-to-event tertiary outcomes, respectively. In accordance with the intent-to-treat principle, the analysis will include all patients in the arm to which they were randomized regardless of study compliance. Based on our substantial prior experience with this population we expect minimal missing data. However, details of missing data will be provided and we will perform a sensitivity analysis using a graphical pattern mixture tipping point approach demonstrating the treatment effect over the possible range of missing outcomes.51,52
What is the frequency of analysis?
Although glutamine is recommended by current guidelines and used in about half of all burn cases, recent safety concerns have emerged.16,17 Therefore, we will test for excess mortality in the glutamine arm after 600 and 1350 patients have been followed for 6 months. These one-sided interim analyses will each be tested at a nominal one-sided P value of 0.01. However, the final assessment after all patients have completed the six-month follow-up will be two-sided. In order to maintain an overall type I error rate of 0.025 in each direction, the final analysis will test for higher mortality in the glutamine arm at a nominal P value of 0.011 while lower mortality in the glutamine arm will be tested at the traditional 0.025. This approach will maintain an overall type I error rate of 0.05 without affecting the power to detect a glutamine benefit. However, the power to detect harm with glutamine will be decreased slightly. We feel this is justified in order to allow the possibility of stopping the study early if a strong signal of increased mortality emerged. Details of and justification for this approach are provided in Appendix 3.
Are there any planned subgroup analyses?
We will perform two pre-specified subgroup analyses based on burn severity (TBSA) and age. The rationale for both of these subgroups is that older patient and patients with more severe burns will likely be deficient in glutamine and therefore more likely to benefit from supplementation.53,54 The statistical significance of apparent effect modification will be assessed by testing a treatment by covariate interaction term using logistic regression for mortality and Cox PH model for TTDA.
Conclusion
This study will be the first large international multicentre trial examining the effects of glutamine in burn patients and the largest study of burn-injured patients ever. Furthermore, it creates a precedent for doing mortality-based trials of burn-injured patients and demonstrates the ability of burn units around the world to work together. Morever, this large dataset of enrolled burn patients may be used to further generate additional knowledge, such as the relationship between nutrition intake and outcome, for exapmle. Negative or positive, the results of this trial will inform the clinical practice of burn care in patients around the world.
Footnotes
Appendix 1: Interim and final analysis boundaries
Despite limited evidence, glutamine is currently a recommended treatment for burn patients and is used in about half of all burn patients. Although equipoise currently exists, glutamine safety concerns have recently arisen in slightly different patient populations. Therefore, we would like to perform an early assessment of increased mortality in the glutamine arm. But given the concerns from other studies, stopping the study early for glutamine benefit may leave lingering uncertainties regarding the safety and efficacy of glutamine in this population. Hence, we propose two one-sided interim analyses to test for increased six-month mortality in the glutamine arm once 600 and 1350 patients are evaluable. True equipoise currently exists, so a study demonstrating increased harm due to glutamine will have as much impact on practice as a study showing a glutamine benefit. Therefore, we have avoided a futility analysis that would not establish if glutamine actually increases mortality. On the other hand, traditional O’Brien-Fleming type boundaries require an unacceptably large increase in mortality to stop the study at the interim analyses. We have chosen a compromise that would stop for increased mortality in the glutamine arm if a nominal P value of 0.01 is reached at the first or second interim analysis. At the first interim analysis, this threshold would be met if the observed six-month mortality rates in the glutamine and control arm were 22.4% and 15.0%, respectively, or 16.4% and 10.0%, respectively. At the second interim analysis, the stopping rule would be met if the observed mortality rates in the glutamine and control arms were 19.8% and 15.0%, respectively, or 14.1% and 10.0%, respectively. In order to maintain the type I error for increased glutamine mortality at 0.025, the final analysis will be performed at a nominal 0.011. The test for glutamine benefit will be performed at completion of the trial at the traditional 0.025. This proposed approach will maintain an overall two-sided type I error rate of 0.05 without affecting the power to detect a glutamine benefit. The slight reduction in power to detect control arm benefit was considered necessary in order to allow for early stopping if unacceptable increase in mortality are observed in the glutamine arm. This approach allows an early assessment of glutamine harm, but will provide a definitive and convincing study conclusion regardless of the direction of benefit.
Tables 1–8 show the power and true type I error rates for the proposed testing strategy under various plausible scenarios. These power and type I error rates were each estimated by one million simulations so that the estimates are accurate to within one-tenth of 1% 19 out of 20 times. Table 1 verifies that the overall type I error rate of the study is 0.05. Table 2 confirms that 80% power is maintained when there is a 25% RR reduction from 15% to 11.25% due to glutamine. Table 3 shows that we have 92% power to detect a 25% RR reduction from 20% to 15% due to glutamine. Tables 4–8 demonstrate the power at each interim analysis under a range of scenarios where glutamine increases mortality.
Appendix 2
Appendix 3. Sample size justification part A: primary outcome – six-month mortality
It may be seen that with a mortality rate of 15% in the placebo arm (red) we achieve 80% power to detect a 25% R reduction (RR = 0.75, experimental mortality rate = 11.25%) or a 30% RR increase (RR = 1.3, experimental mortality rate = 19.5%). Our power will increase if the prior literature showing higher mortality rates is true.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: A.F.T. is the Canada Research Chair in Critical Care Neurology and Trauma.
Funding
The RE-ENERGIZE study has been funded by the Canadian Institutes of Health Research. The study products have been provided by Emmaus Inc.