
Abstract
Several studies have shown that gram-negative bacilli infection can cause acute lung injury, and that consequent pulmonary fibrosis is caused when alveolar type-II epithelial cells undergo epithelial-mesenchymal transition (EMT). However, the mechanism underlying this change remains unclear. This study aimed to elucidate whether the main toxin of gram-negative bacteria, lipopolysaccharide (LPS), can induce EMT in human alveolar epithelial cells, and the underlying molecular mechanisms. Human alveolar type-II epithelial cells (A549) were used in EMT induction experiments. Cells were collected after LPS exposure, and changes in the expression levels of epithelial and mesenchymal cell markers were determined. Further, the effect of LPS exposure on the expression of Toll-like Receptor 4 (TLR4), Transforming Growth Factor-beta 1 (TGF-β1) and Smad2/3 was assessed. The expression level of a mesenchymal cell marker was also assessed after pharmacological inhibition of TLR4 and TGF-β1 prior to addition of LPS, to identify downstream pathways involved in EMT induction. Results showed that LPS exposure caused significant downregulation of epithelial marker E-cadherin, and upregulation of mesenchymal marker vimentin, together with increased expression of TGF-β1 and activation of the TGF-β1/Smad2/3 pathway. Furthermore, pretreatment with TGF-β1 and TLR4 inhibitors suppressed EMT, and treatment with the latter also reduced the expression level of TGF-β1. Overall, we conclude that LPS directly induces EMT in A549 cells through upregulation of TLR4 and activation of the TGF-β1/Smad2/3 signalling pathway. Our results suggest that LPS-mediated pulmonary fibrosis may occur in ALI patients even if the LPS-induced inflammatory response is inhibited.
Keywords
Introduction
Acute lung injury (ALI), characterised by acute hypoxic respiratory insufficiency as the main clinical manifestation, is caused by various pathogenic factors, resulting in diffused pulmonary interstitial and alveolar oedema. In the long term, ALI may be related to fibroblast and myofibroblast proliferation and new matrix deposition.
In the clinic, gram-negative bacilli infection is an important cause of ALI. Lipopolysaccharide (LPS), the main toxin of gram-negative bacilli, has been used in many in vivo studies to establish pulmonary fibrosis models.1–6 It has been demonstrated that LPS binds to its cognate partner Toll-like receptor 4 (TLR4),7–10 and that activation of the TLR4 signalling pathway is critical for induction of fibrosis.11–13 Activation of the TLR4 pathway in prostate epithelial cells leads to increased Transforming Growth Factor Beta 1 (TGF-β1) expression.14–16 Moreover, TGF-β1/Smad has been proven to be an important pathway leading to epithelial-mesenchymal transition (EMT).17–20
Although the specific mechanism of pulmonary fibrosis remains unclear, the most widely recognised hypothesis for the pathogenesis of pulmonary fibrosis relies on the insufficient ability of the alveolar epithelium to regenerate after injury. Indeed, when alveolar epithelial cells become apoptotic and insufficient to renew, fibroblasts and myofibroblasts accumulate and deposit extracellular matrix. Despite controversy, EMT, may be one of the main forces driving this process. 21 Given that EMT may play a key role in the process of pulmonary fibrosis, the aim of this study was to elucidate whether the main toxin of gram-negative LPS can induce EMT in a human alveolar type-II epithelial cell line, and explore the possible signalling pathways.
Materials and methods
Cell culture
A549 cells were purchased from the Peking Union Cell Resource Center (Beijing, China). Cells were routinely cultured in an RPMI 1640 medium (Gibco, NY, USA), containing inactivated 10% foetal bovine serum (Hyclone, Utah, USA) and supplemented with 100 U/mL penicillin and 100 µg/mL streptomycin (Gibco, NY, USA), in an incubator maintained at 37°C and 5% CO2.
Western blotting
Cells were stimulated as described above, harvested and then lysed using a buffer containing NP-40 (Solarbio, Beijing, China) for total cellular protein extraction. Equal amounts of protein were separated using sodium dodecyl sulphate-polyacrylamide gel electrophoresis and subsequently transferred onto polyvinylidene difluoride membranes (Millipore, Milan, Italy). The membranes were incubated overnight at 4°C with appropriate primary antibodies against E-cadherin (1:200, Cell Signaling Technology, USA), vimentin (1:200, Cell Signaling Technology, USA), p-Smad2/3 (1:200, Cell Signaling Technology, USA) and β-actin (1:200, ZSGB-BIO, China) was used as loading control. The following day, the membranes were incubated with suitable horseradish peroxidase-linked secondary antibodies (1:3000, Earthox, USA), and the protein bands were visualised using an enhanced chemiluminescence kit (Sangon Biotech, Shanghai, China).
Real-time reverse-transcriptase PCR (qRT-PCR)
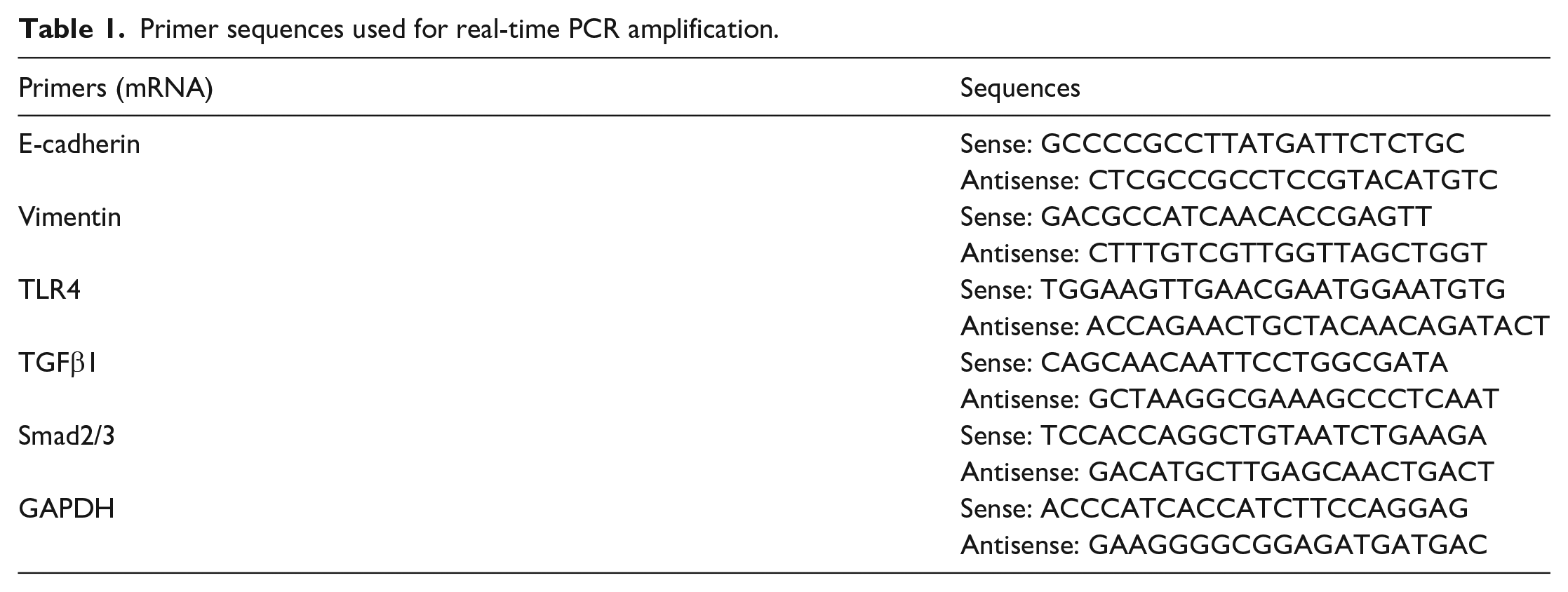
Total RNA was extracted from cells using Trizol Reagent (Thermo Fisher, USA), and RNA quality was determined using an Eppendorf Bio Photometer (Hamburg, Germany). cDNA was synthesised using the isolated RNA as template, using Synthesis Kits according to manufacturer’s instructions (Tiangen, Beijing, China). The primers used for amplification were provided by Sangon Biotech (Shanghai, China) and are listed in Table 1. Quantitative PCR analyses were carried out to detect mRNA expression using the PowerUp SYBR Green Master Mix (Thermo Fisher Scientific). GAPDH was used as an internal control.
Primer sequences used for real-time PCR amplification.
Statistical analysis
Data are presented as mean ± SD from at least three independent experiments. Unpaired t-tests were used for pairwise comparisons, and ANOVA with Dunnett’s post-test for multiple sets of data. A value of p < 0.05 was defined as significant.
Results
Lipopolysaccharide induces epithelial-mesenchymal transition (EMT) in A549 cells in a TGF-β1/Smad2/3 pathway-dependent manner
To investigate whether LPS directly induces EMT without the involvement of immune cells, secondary factors and other mediators, alveolar epithelial cells (A549) were stimulated with LPS(net concentration 10 µg/mL) for 0.5, 1, 2, 6 and 12 h, and the levels of mRNA of genes associated with EMT were examined with qRT-PCR. It was observed that the mRNA level of epithelial marker E-cadherin gradually decreased with prolonged LPS stimulation (Figure 1(a)). In contrast, the mRNA level of mesenchymal marker vimentin increased significantly after 1 and 2 h of LPS stimulation (p < 0.05; Figure 1(b)). Similarly, the expression of TGF-β1 mRNA peaked 1 h after LPS stimulation and remained high thereafter, in contrast to the control group (p < 0.05; Figure 1(d)). The mRNA level of Smad2/3 was markedly increased at 1 h after LPS stimulation (p < 0.05; Figure 1(e)). The results of qRT-PCR experiments were corroborated with western blot analysis performed on whole cell lysates after treatment with LPS (Figure 2(a)–(c)). Activation of the Smad2/3 signalling module was checked by probing for phosphorylated Smad2/3 (p-Smad2/3), which was observed 2 h onwards of LPS treatment (Figure 2(a) and (d)).

Temporal changes in mRNA expression levels of EMT-related markers in A549 cells upon LPS treatment. LPS (net concentration 10 µg/mL) was added to A549 cells, and real-time PCR was used to measure the mRNA levels of EMT-related markers 0.5, 1, 2, 3, 6 and 12 h after LPS treatment. The epithelial marker E-cadherin mRNA decreased significantly compared to the untreated control group at each time point (a), while the mesenchymal marker vimentin mRNA increased significantly 2 h after LPS stimulation (b). The expression of TLR4 mRNA increased 1 h after LPS treatment (c) and that of TGF-β1 mRNA significantly increased 1, 2, 6 and 12 h post LPS treatment (d). Smad2/3 mRNA expression also significantly increased 1 h after LPS treatment (e).

Protein expression changes in EMT-related markers in A549 cells after LPS treatment. Western blot showing increase in the expression of E-cadherin compared to untreated control cells after LPS (net concentration 10 µg/mL) stimulation of A549 cells for 1, 6 and 12 h (a, b). Expression of vimentin increased significantly compared to that in the control group at each time point (a, c), and expression of p-Smad2/3 protein detected 0.5, 1 and 2 h after LPS stimulation was also significantly increased compared to that in untreated control cells (a, d).
To investigate whether LPS-induced EMT was occurring through the TGF-β1 pathway, we exposed A549 cells to the TGF-β1 inhibitor EW7197 (10 µM, Selleck Chemicals, USA) prior to treatment with LPS and analysed protein expression levels of EMT-associated markers, using Western blot (Figure 3(b) and (d)). The results showed that compared with the LPS-stimulated group, pretreatment of cells with EW7197 0.5 h before addition of LPS significantly reduced vimentin protein expression, suggesting that the TGF-β1 pathway plays an important role in the EMT induced by LPS stimulation. To summarise, the above data indicate that LPS induces EMT in A549 cells, likely in a TGF-β1/Smad2/3 pathway-dependent manner.

Quantification of changes in vimentin protein level after inhibition of TGF-β1 and TLR4 prior to stimulation by LPS. A549 cells were treated with LPS (10 µg/mL) with or without pretreatment with the TLR4 inhibitor TAK242 (10 µM) or the TGF-β1 inhibitor EW7197 (10 µM) for 0.5 h. Pretreatment of A549 cells with inhibitors targeting TLR4 (a, c) and TGF-β1 (b, d) before LPS stimulation significantly lowered the expression of vimentin.
Lipopolysaccharide-induced epithelial-mesenchymal transition is mediated by TLR4
In order to detect changes in mRNA expression levels of TLR4 upon stimulating alveolar epithelial cells with LPS (10 µg/mL), qRT-PCR was performed on samples collected 0.5, 1, 2, 6 and 12 h after LPS stimulation. The results showed that compared to the untreated control group, the TLR4 mRNA expression level in the LPS-treated group significantly increased 1 h after LPS treatment (p < 0.05; Figure 1(c)). To further investigate whether the TLR4 pathway played a role in EMT, we pharmacologically inhibited TLR4 in A549 cells, using the small molecule TAK242 (10 µM, Selleck Chemicals, America), for 0.5 h prior to LPS treatment for 1 h and examined corresponding effects on EMT. Pretreatment of A549 cells with TAK242 before LPS exposure significantly reduced vimentin expression compared to that after LPS stimulation alone (Figure 3(a) and (c)). Inhibition of TLR4 also dramatically reduced TGF-β1 expression, as assessed with qRT-PCR (Figure 4).

TGF-β1 expression level upon pharmacological inhibition of TLR4 prior to LPS stimulation. A549 cells were pretreated with TAK242 (10 µM) for 0.5 h before stimulating with LPS (10 µg/mL) for 1 h; the levels of TGF-β1 mRNA were significantly reduced compared with those in the untreated control group.
Discussion
ALI is characterised by diffused pulmonary interstitial and alveolar oedema, which leads to acute hypoxic respiratory insufficiency and pulmonary fibrosis, posing a significant health threat to individuals.22,23 However, the precise molecular mechanism behind the development of pulmonary fibrosis after ALI remains unclear. During epithelial injury repair, alveolar type II epithelial cells act as stem cells and undergo transdifferentiation to alveolar type I epithelial cells, thus ensuring the renewal of the latter. However, studies have found that in some cases, alveolar type II epithelial cells lose their regenerative potential and differentiate into mesenchymal cells, which contributes to the pathogenesis of pulmonary fibrosis, and EMT may be one of the major driving forces in the process.21,24 Considering that EMT mechanism may play an important role in lung fibrosis after lung injury, we aim to elucidate whether the main toxin of gram-negative LPS can induce EMT in a human alveolar type-II epithelial cell line, and explore the possible signalling pathways.
In clinical practice, gram-negative bacilli infection is an important cause of ALI. 25 Previously animal models have been used to show that LPS of gram-negative bacteria is a potent inducer of pulmonary fibrosis after ALI4,6 However, these studies did not address whether LPS could directly induce pulmonary fibrosis or if this phenomenon could have been a consequence of LPS-regulated changes in other lung cells and inflammatory factors. Our study demonstrates that LPS can directly induce the conversion of alveolar type II epithelial cells to mesenchymal cells in vitro without the involvement of other pro-inflammatory cells or factors. In the model system of A549 cells, expression of the epithelial cell marker E-cadherin decreased, while that of the mesenchymal cell marker vimentin increased 1 h after LPS stimulation, at which time cells are likely transitioning from epithelial cells to fibroblasts. Therefore, our results suggest that if epithelial cells cannot avoid contact with LPS, pulmonary fibrosis may still occur, even if the pulmonary inflammatory response caused by LPS during pulmonary fibrosis treatment is inhibited.
Our results showed that treatment of alveolar epithelial cells with LPS increased the expression levels of EMT-associated genes together with TGF-β1 and Smad2/3, and activated the TGF-β1/Smad2/3 signalling pathway, as evidenced by detection of p-Smad2/3 signal in LPS-stimulated cells. Indeed, previously published studies have also shown that activation of the TGF-β1/Smad2/3 pathway is an important pathway that promotes EMT.17–20 Interestingly, LPS-induced EMT was significantly inhibited when cells were pretreated with TGF-β1 inhibitor, suggesting that LPS causes EMT by upregulating TGF-β1, thereby activating the TGF-β1/Smad2/3 pathway.
How does LPS increase TGF-β1 levels? The current study shows that the expression of TLR4, the binding partner of LPS on the cell membrane, was upregulated after LPS treatment. Further, expression of TGF-β1 and consequent EMT was also significantly suppressed when TLR4 was inhibited, suggesting that TLR4 is potentially a key mediator in the crosstalk between LPS and the TGF-β1/Smad2/3 pathway. It is conceivable that engagement of TLR4 by LPS triggers an intracellular signalling cascade that leads to activation of the TGF-β1/Smad2/3 pathway that finally leads to EMT and favours excessive deposition of extracellular matrix, which eventually causes fibrosis. Figure 5 represents a schematic diagram of the process of LPS-induced EMT in our study. In addition, other epigenetic factors may be involved in LPS-induced EMT. For example, micro-RNA (miRNA)-200 family members have been found to exert a protective effect on pulmonary fibrosis by targeting ZEB1/2 via the p38/MAPK and TGF-β/Smad3 signalling pathways, 26 and by enhancing IPF alveolar type II epithelial cell transdifferentiation into type I epithelial cells to rescue pulmonary fibrosis. 27 Thus, different epigenetic patterns may explain why only a few cases of ALI accompanied by gram-negative bacteria infection progress to pulmonary fibrosis.

Schematic diagram of EMT induction by LPS in alveolar epithelial cells. Schematic diagram of LPS-induced changes in A549 cells. Arrows represent activation, T-heads represent inhibition.
It must be highlighted that despite some progress in research on inflammation and immunotherapy, the precise mechanism underlying the pathogenesis of pulmonary fibrosis is still unknown. 28 Moreover, pathobiological processes occurring in A549 cells during LPS-induced EMT may not model disease progression in humans. A549 cells are derived from non-small cell human lung cancer cells, which are indeed different from healthy lung cells and in vivo studies. In addition, the concentration of LPS used in in vitro approaches is often much higher than that observed physiologically.
Therefore, our research provides a reference for future studies, which will need to reproduce this mode of EMT induction in healthy alveolar epithelium or in other physiologically relevant model systems and explore underlying molecular mechanisms.
Conclusions
Our study demonstrates that LPS can directly promote the transition of alveolar type II epithelial cells to mesenchymal cells without the involvement of inflammatory cells, factors, and secondary mediators, which greatly increases the risk of developing pulmonary fibrosis. Acquisition of mesenchymal features by alveolar type II epithelial cells is mediated by upregulation of the LPS receptor TLR4 and activation of the TGF-β1/Smad2/3 signalling pathway. Our results suggest that pulmonary fibrosis may occur via the aforementioned mechanism in ALI patients suffering from gram-negative bacilli infection, despite clinical inhibition of the LPS-induced inflammatory response. Thus, our study sheds light on strategies to improve the therapeutic intervention of pulmonary fibrosis following ALI caused by gram-negative bacilli. However, despite this finding in A549 cells, more work should be done to reproduce this mode of EMT induction in healthy alveolar epithelium or in other physiologically relevant model systems and explore underlying molecular mechanisms.
Footnotes
Author contributions
Shaui Wu designed and carried out experiments, collected and analysed data, created pictures and tables and wrote the paper; Tian Jiao Xue and Jiali Wang helped in conducting experiments, data analysis and manuscript editing; Huan Ye designed the entire study, organised data and significantly revised the manuscript.
Availability of data and materials
The data and materials used during the study are available from the corresponding author upon reasonable request.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval
Ethical approval was not sought for the present study because the A549 cell line used in our in vitro research is a commercial product specially produced for scientific research, which has been widely used for more than 40 years. Any professional engaged in scientific research can buy it from formal channels, so there is no ethical crisis in our research.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Science and Technology Program Funding Project of the Beijing Municipal Education Commission (KM201710025017). Our research design, data analysis and manuscript writing were independent of the funding.