
Abstract
The public health crisis of the novel coronavirus disease (COVID-19) is alarming since January 2020. COVID-19 genome (SARS-CoV-2) is related to other highly pathogenic coronaviruses SARS-CoV (severe acute respiratory syndrome coronavirus) and MERS-CoV (Middle East respiratory syndrome coronavirus). Amino acid substitutions in some of SARS-CoV-2 proteins resulted in mutations proposing more virulent and contagious properties for this novel virus. Coronavirus penetrates the host cell via endocytosis and once infected, immune responses are triggered to fight against the pathogen. Innate immune response activates major transcription factors to secrete proinflammatory cytokines and type 1 interferon response (T1INF) to induce antiviral immunity. While adaptive immunity initiates cascade of B-cells antibody mediated and T-cells cellular mediate immunities, several mechanisms are raised by SARS-CoV-2 to evade host immune response. Consequently, a surge of proinflammatory cytokines, known as cytokine storm (CS) are released. Failure to manage CS results in several pathological complications as acute respiratory distress syndrome (ARDS). Although researches have not discovered an effective treatment against SARS-CoV-2, recent therapeutic approaches recommending the use of anti-inflammatories in combination with antivirals and some repurposed drugs for COVID-19 patients. Future medications should be designed to target essential hallmarks in the CS to improve clinical outcomes.
Background
In December 2019, the global pandemic of novel coronavirus (nCoV) disease (COVID-19) believed to be started in Wuhan, China, has subsequently dispersed around the globe 1 leading to the death of more than one million people worldwide. 2 Cytokine storm (CS) occurs when the immune system is exaggerated after SARS-CoV-2 infection, and it appears to be one of the common causes of mortality in the recently declared pandemic of COVID-19. 3 Therapeutic approaches to manage CS might provide an avenue to decrease the COVID-19 associated morbidity and mortality rates. 4 Moreover, drug repurposing has been approved to be effective with COVID-19 patients. 5 The aim of this mini-review is to provide a comprehensive knowledge on COVID-19 infection and inflammatory pathways, in addition to highlight the importance of managing CS and the different treatment options associated with COVID-19 pandemic.
Pandemic challenge
As the world is facing the COVID-19 outbreak; a disease originated from the new coronavirus SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2), clinical data, and emerging genetics indicate a similar route to SARS-CoV and MERS (Middle East respiratory syndrome coronavirus) diseases. Merging advanced vaccine technology, high throughput genomic sequencing and open access datasets are expected to provide more knowledge about this pathogen, including the host immune response, as well as the plan for possible future therapeutics strategies. 6
Coronaviruses infecting humans are classified into two major classes: low pathogenic and highly pathogenic CoVs. Low pathogenic human coronaviruses (HCoV) such as HCoV-229E, HCoV-NL63, HCoV-HKU, and HCoV-OC43 infect the uppers airways causing occasional mild to severe cold-like respiratory diseases. While, highly pathogenic HCoVs including SARS-CoV and MERS-CoV 7 invade the lower respiratory tract, causing pneumonia that may result in catastrophic acute lung injury (ALI) and acute respiratory distress syndrome (ARDS).8–10 Infection with highly pathogenic HCoVs causes wide range of clinical symptoms, where the majority of patients have phases of mild clinical illness, and only a limited number face serious ALI and ARDS.11,12
SARS-CoV, MERS-CoV, and SARS-CoV-2 are categorized as family members of the beta-coronavirus. 13 SARS—like coronaviruses originated in bats, while MERS-CoV have transferred from camels, make it possible that such viruses may begin to cross barriers between organisms and trigger more outbreaks in human populations.14,15 A recent study found out a strong genetic homology between SARS-CoV-2, MERS-CoV, and SARS-CoV. 16 The first recorded case of SARS-CoV infection in Guangdong Province of China was retrospectively reported in November 2002. SARS-CoV outbreak infected over 8400 confirmed cases in 37 countries over a period of 7 months, with a disease gross mortality rate of 9.6%. 17 Recently in April 5, 2017, MERS-CoV infected 1936 individuals and caused death to 690 patients. 18 As for February 6, 2021, World Health Organization (WHO) reported 104,956,439 confirmed cases of COVID-19, including 2,290,488 death cases. 2
Viral genome
Coronaviruses are ribonucleic acid (RNA) viruses which are single-stranded, positive-sense, and enveloped nucleic acid. Genomes have a 5′ cap and 3′ poly (A) tail and are classified into non-structural protein genes and protein genes of structural and accessory type. The main structural proteins include envelope (E), matrix (M), spike (S), and nucleocapsid (N) proteins. 19 Spike protein cleavage is essential for the initiation of host-virus membrane fusion and eventually cell entry. SARS-CoV-2 enters the cells when the viral structural S protein binds to the angiotensin-converting enzyme 2 (ACE2) receptor, thus virus particle enters the cell through its receptors and endosome formation.20,21 In general, RNA viruses have intrinsically higher rates of mutation than deoxyribonucleic acid (DNA) viruses, due to the absence of proofreading ability in RNA-dependent polymerase. 22 This enables them to develop at a rapid pace, particularly in combination with massive population sizes and short-term generation times. Indeed, RNA viruses have been responsible for several viral emergencies in the human communities over the past few decades, 23 such as Hepatitis C virus causing chronic hepatitis and it is an important risk factor for hepatocellular carcinoma progression, Ebola virus disease caused by Ebola virus, polio measles occurred after getting infected by poliovirus, and Orthomyxoviruses (influenza viruses) resulting in influenza disease. Other RNA viruses including Human immunodeficiency virus that causes Acquired immunodeficiency syndrome in its late stages, and Human T-cell leukemia virus (HTLV-1) that develops adult T-cell leukemia and HTLV-1 associated myelopathy/tropical spastic paraparesis.24–27
Coronaviruses are unique RNA viruses possessing proofreading abilities in their genomes. Non-structural protein (nsp) 14 labeled as ExoN, interacts with nsp10 to perform powerful exoribonuclease activity from 3′–5′. This behavior is comparable to the DNA polymerase proofreading behavior, 28 as expressed in nsp14 by conserving the DEDD superfamily motif, a characteristic exonuclease activity among DNA species. If this motif is mutated in nsp14, the virus will have a 15–20 fold rise in the rate of mutation. 29 The improved reliability of the nsp10–nsp14 complex and the subsequent lower mutation rate seem to have granted wild-type coronaviruses to avoid error and extend their genomes by twice the fold size of the next largest genomes of RNA viruses. 30
SARS-CoV-2 in terms of the whole genome sequence is similar to the SARS-like bat CoVs. Most SARS-CoV-2 genomic encoded proteins share similarity with SARS-CoVs proteins along with some variations. At the protein stage, there are no substitutions of amino acids in NSP7, NSP13 envelope, matrix or accessory proteins p6 and 8b, except for NSP2, NSP3, spike protein, subdomain underpinning that is, receptor- binding domain (RBD). 31 Another recent work suggests that NSP2 and NSP3 mutations play a role in SARS-CoV-2’s infectious capacity and differentiation process. This allows researchers to investigate the gap between SARS-CoV-2 and SARS-CoV in the host tropism and transmission, and to perform further studies on possible therapeutic targets. 32 Other CoVs existed in different animal species; including birds and mammals, are classified as alpha, beta, gamma, and delta coronaviruses.33,34
Three stages were observed in SARS-CoV-2 infection affecting patients differently. Stage I, asymptomatic phase of incubation with or without observable virus in infected patients’ blood, stage II, non-serious symptomatic cycle with detectable virus titer, and stage III, severe respiratory symptomatic stage with high viral content detected in infected patients. 35 Earlier it was proposed that SARS-CoV-2 would be conveyed from an asymptomatic carrier to near contacts, although this has been questioned. However, even if family members and near contacts may be compromised by the index patient during the pre-symptomatic window duration as stated, a serious concern should be addressed. 36 A study reported that SARS-CoV-2 had mutated in specific patients in China, although its degree of mutation is smaller than that of H7N9 avian influenza.33,34 A community genetic study was conducted on 103 SARS-CoV-2 genomes that identified two forms of SARS-CoV-2, L type (~ 70%) and S type (~ 30%). The L strains originating from the strain S are more aggressive and infectious. The latest coronavirus needs further examination for better understanding to its epidemic nature and virulence. 34
The research nowadays is focused on identifying novel targets to facilitate SARS-CoV-2 recognition. One example is the heterodimer S100A8/A9, which is constituted of two Ca2+ binding proteins; S100A8 (MRP8) and S100A9 (MRP14). 37 S100A8/A9 is expressed as a Ca2+ sensor in the neutrophils and monocytes, and it plays a major role in moderating the inflammatory response via leukocytes recruiting and cytokines release. Such heterodimer could serve as a diagnostic biomarker in the inflammation-associated diseases, while S100A8/A9 inhibitors could be used as potential therapeutic targets in inflammatory conditions.38,39 Another target is the high mobility group box-1 (HMGB1); a chromatin-linked protein that possess cytokine activity. HMGB1 was reported to stimulate the leukocyte adhesion, enhance the release of proinflammatory cytokines and the production of inflammatory mediators as angiogenic factors. 40 Both S100A8/A9 and HMGB1 serum levels are upregulated in COVID-19 patients, and it was observed that their elevated serum levels are associated with severity of the tissue damage and the excessive release of cytokines. Thus, they are considered indicators for the disease progression and alarming sign for the clinical risks and fatal outcomes of COVID-19. 38
Moreover, understanding the exact mechanism will enable identifying plethora of targets to develop antiviral drugs or repurpose the use of others. 41 Researchers rely on the use of computational biology and in silico screening to identify potential targets. Four commercial drugs showed promising inhibition of main protease dimer (Mpro) and transmembrane protease serine 2 (TMPRSS2) enzymes. 42 Human immunodeficiency virus (HIV) protease inhibitors were suggested as one of the potential therapies using in silico studies. 43 Furthermore, glecaprevir, paritaprevir, simeprevir, ledipasvir, glycyrrhizic acid, TMC-310911, and hesperidin showed promising effect in their free binding energies with all tested target proteins. 44
SARS-CoV-2 infection
SARS-CoV-2 invades the host cell through binding of viral spike protein to host’s ACE2 cell receptor as previously mentioned. Although ACE2 is expressed in the majority of the body organs (including lungs, oral, and nasal mucosa, nasopharynx, skin, stomach, small intestine, brain, etc.), 45 it is highly expressed on lung alveoli, secretory goblet and ciliated epithelial cells.46,47 A recent research examined SARS-CoV-2 infected cultures, reported that the virus is targeting the ciliated cells. This result was proved by colocalization of the viral capsid S protein with acetylated tubulin, a marker for the ciliated cells. 48 Another study reported a three-fold increase in ACE2 expression in a group of epithelial cells after infection with SARS-CoV-2. Infected epithelial cells trigger the release of chemokines and stimulate host immunity to attract immune cells (e.g. neutrophils, cytotoxic T-cells, CD4 T-cells, T regulatory cells, and mast cells) at the infection site. 49 SARS-CoV-2 replication is extended from the nasopharynx to the lower respiratory tract, which may contribute to its rapid transmission. 50 Both SARS coronaviruses share some similarity in the infection pathophysiology, 51 although the aggressiveness of SARS-CoV-2 is attributed to the severe inflammatory responses and the massive tissue damage. Subsequently, the host response is a key player in the disease intensity along with viral load. 52 Once the body is exposed to SARS-CoV, the most infected immune cells are monocytes/macrophages and T cells. Similar mechanism is proposed for SARS-CoV-2 host immune interaction, as these cells were shown to have ACE2 receptors on their surfaces.53,54 In the following section, we are going to discuss the host immune responses and different mechanisms by which SARS-CoV-2 can escape such immune response.
Host immune response
Host immune defense to viral infection is managed through immune stimulation and programmed cell death. 55 Both the adaptive and the innate immunity are joined to destroy the virus. 56
Innate immunity
The first defense line encountering any invading pathogen is the innate immunity. The main target of the innate immune response is blocking the propagation and replication of the pathogen within the body immediately after the infection. 57 It is considered as non-specific type of immunity, which recognizes conserved molecules present only in the pathogen and absent in the host, so that it can effectively fight the invading microorganism without damaging the host. These microorganism-associated features (known as pathogen-associated immunostimulants) can induce two pathways by which the innate immune response acts: phagocytosis via neutrophils and macrophages as well as systemic inflammatory responses. Both responses start immediately after the infection without any need to previous exposure history to the microorganism. 58
Immune cells can recognize the viral infection through the detection of virus derived pathogen associated molecular patterns (PAMPs) (e.g. viral RNA). Consequently, this stimulates pattern recognition receptors (PRRs) located inside or on the surfaces of immune cells resulting in immune cells stimulation. Because SARS-CoV-2 is an RNA virus, it is sensed by the immune cells through endosomal RNA PRRs; such as Toll-like receptors (TLR-3 and 7), in addition to some cytoplasmic RNA sensors; including melanoma differentiation-associated protein five (MDA5) and retinoic acid-inducible gene I (RIG-I) (Figure 1). 53 Activation of TLR3/7 affect major transcription factors as nuclear factor Kappa (NFκB) and interferon regulatory factor three (IRF3) causing their transportation into the nucleus54,59 while activation of MDA5/RIG-1 triggers IRF3 activation. Thus, NFκB triggers the release of innate proinflammatory cytokines; including interleukins (IL-1/6) and tumor necrosis factor alpha (TNF-α), while IRF3 stimulates type 1 interferon (T1IFN) expression. When T1IFN is expressed, cellular response is reprogrammed to the anti-viral state, enhancing infection control, and promoting viral clearance.60,61
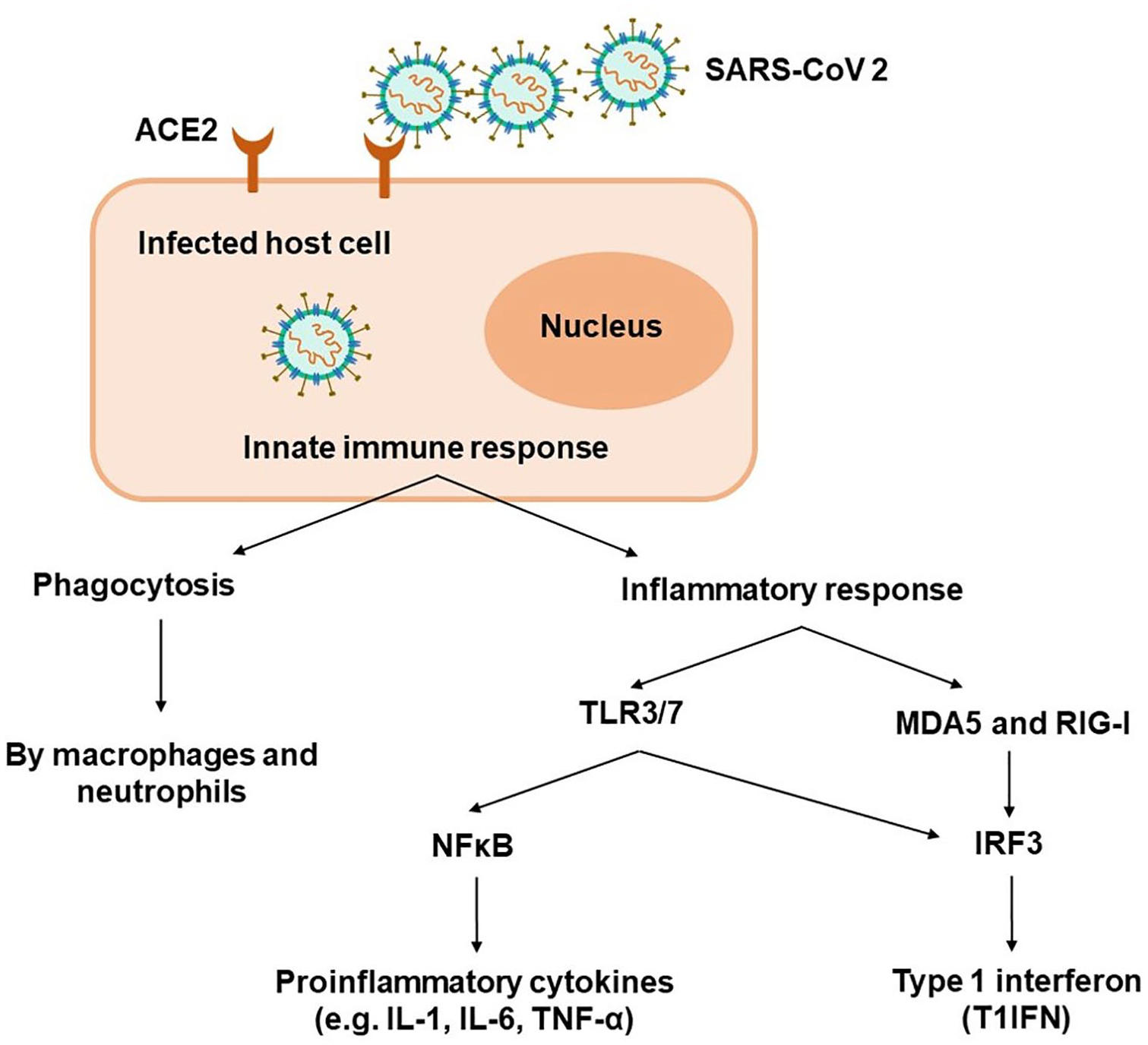
Activation of innate immune response. SARS-CoV-2 enters the host cell via ACE2 receptor on the cell surface. Viral infection stimulates host’s innate immunity through phagocytosis and activation of inflammatory responses. The cell detects viral infection via endosomal RNA toll-like receptors (TLR3/7) that activates NFκB to releases pro-inflammatory cytokines and IRF3 transcription factors. While viral recognition through cytoplasmic RNA sensors MDA5 and RIG-I activates only IRF3 resulting in triggering type 1 interferon.
Adaptive immunity
Adaptive immune response; which is called the specific immunity, is activated secondary to the innate immunity upon stimulation with certain pathogen. Adaptive immune cells sustain long life immunity against the invading microorganisms. The importance of such immunity is highlighted by its ability to destroy pathogens and clear the body from their toxic products. 62 Similar to the innate immunity, the adaptive immune cells attack only the foreign particles not the host. Viral infection triggers antigen-specific immune response, 63 with two main players B-cells and T-cells that mediate two classes of adaptive immunity: antibody response (humoral immunity) and cell mediated immune response respectively. 64
The adaptive immune response is initiated when an antigen presenting cell (APC) activates naive T-cells by displaying the antigen, as T-cells cannot bind to free antigens in the body (Figure 2). 65 Any viral-infected cell triggered to express molecules of major histocompatibility complex (MHC) class I on its surface, can act as APC. 66 Professional APC as dendritic cells, B-cells, and macrophages can engulf the virus by endocytosis, digest its viral material and use small peptide fragments as an antigen on the MHC class II binding cleft present on the surface of APC. 66 T-cell receptor (TCR) located on the surface of naive T cell, can identify the conformational structure of peptide antigen bound to cleft of MHC on APC causing its activation. Thus, major histocompatibility complex antigen loci called human leukocyte antigen (HLA), plays essential role in the genetic susceptibility of infectious diseases.67,68 Different haplotypes are strongly correlated to disease susceptibilities according to previous studies reported the association between specific HLA haplotypes and influenza viruses (such as H1N1). 69 Consequently, identifying alleles of MHC class I or II, in addition to HLA loci correlated with SARS-CoV-2, would provide better explanation to anti-SARS-CoV-2 immunity and facilitate development of suitable clinical management strategies.

Activation of adaptive immune response. Antigen presented on the antigen presenting cell (APC) triggers naive T-cell to initiate adaptive immune response. Humoral immunity is activated through B-cells which release antibodies responsible for viral clearance. Stimulation of cell mediate immunity is achieved via activation of T-cell. CD4+ T-cells activates T-helper (Th) cells to release cytokines, stimulates CD8+ cells to damage the viral infected host cell, and possess stimulatory role in antibodies secretion via B-cells.
Generally, in response to pathogen infection, B-lymphocytes direct antibody mediated immune response to neutralize the virus through the release of a type of antibodies (Ab) called immunoglobulins, that recognize foreign antigen (Ag) (the virus and its toxic products). Binding of immunoglobulins to their specific antigen prevents the virus from binding to host’s cell surface receptor, in addition to marking the virus for phagocytic ingestion by the innate immune cells. 70 Moreover, cell mediate immune response is induced when stimulated T-cells initiate proliferation, differentiation, and production of cytokines. 71 Activated CD4+ T-lymphocytes act as T-helper (Th) cells, which release proinflammatory cytokines to eliminate the virus and help other T-cells (Figure 2). They trigger B-lymphocytes to release virus-specific antibodies, 72 and CD8+ T-lymphocytes to develop cytotoxic T-lymphocytes, which identify the viral infected host cell via viral antigens on the cell surface, directly attack and lyse the infected host cell. 70
However, SARS-CoV-2 stimulates destruction and death of the virus infected cells during the virus replication cycle as a part of its cytopathic mechanism. 73 Viral infection could also induce certain inflammatory pathway called pyroptosis, characterized by programmed cell death, normally detected with cytopathic viruses.74,75 Pyroptosis induces the release inflammatory cytokines for example, IL-1β, in addition to other proinflammatory cytokines and inflammatory chemokines such as IL-6, interferon gamma (IFNγ), interferon gamma-induced protein 10 (IP-10), and Monocyte chemoattractant protein-1 (MCP1), which are found elevated in patients with SARS-CoV-2.76,77 Consequently, immune cells such as monocytes and T-lymphocytes are recruited from the circulation to the site of infection.78,79 Pulmonary attraction of such immune cells and the infiltration of the lymphocytes in the respiratory airway are the main reasons of lymphopenia and elevated ratio of neutrophil to lymphocytes in SARS-CoV-2 patients’ blood. 52
SARS-CoV-2 infection is correlated with dysregulation in the immune system both quantitatively and qualitatively. Analysis of COVID-19 patients peripheral blood revealed hyperactivation of lymphocytes, followed by significant reduction in total lymphocytes count. B-cells, CD4+ and CD8+ T-cells total counts were lower in patients with advanced stages when compared to mild cases. 80 CD8+ T-cells concentrations were inversely correlated with some inflammatory indicators such as IL-6, C-reactive protein (CRP), and erythrocyte sedimentation rate (ESR), while the association was positive in case of CD4+/CD8+ ratio. CD4+ T-cells concentration and total lymphocytes counts were inversely correlated with ESR, while natural killer (NK) cells were in negative association with IL-6. 81 The marked lymphopenia in COVID-19 patients 82 may be due to the excessive T-lymphocyte activation, and the increase in CD8+ and Th17 cells secondary to viral infection, resulting in massive destruction in the immune system and lymphocytes depletion. 83 Another potential explanation is that lymphocytes have plenty of ACE2 receptors on their surfaces, marking them for viral attachment and early destruction. 84 Qualitatively, neutrophil to lymphocyte ratio was elevated and the functions of CD8+ and NK cells were impaired in patients with SARS-CoV-2. 85 Therefore, lymphocytes might act as predictor for the infection severity, disease progression, and prognosis. 86
For better interpretation of the immune responses, some studies examined different immunological markers in SARS-CoV-2 patients’ peripheral blood. A recent research identified immune signature to differentiate between COVID-19 patients and other patients with different lower respiratory tract infection or healthy individuals. They reported that COVID-19 patient’s blood is boosted with SARS-CoV-2-specific antibodies, highly cycling T-cells, and CD8+ T-cells, which release exhaustion-associated markers, chemokine IP-10, cytokines IL-8, IL-6, and IL-10, and plasmablasts. They also reported that αβ and γδ T-cells were consumed, dendritic cells, and B-cell compartment compositions were changed. 87 Moreover, another study performed single-cell RNA sequencing for peripheral blood mononuclear cells (PBMCs) collected from COVID-19 patients and patients infected with influenza A virus. They discovered increase in X-linked inhibitor of apoptosis (XIAP)-associated factor 1 (XAF1), TNF, and FAS-induced T-cell apoptosis in SARS-CoV-2 patients. Additionally, they highlighted specific signaling pathways as STAT1 and IRF3 that were activated in COVID-19 patients. Consequently, IL-6R, IL6ST, and proinflammatory cytokines expressions were elevated. These data revealed definite immune response pathways in case of SARS-CoV-2 infection. 88
SARS-CoV-2 evasion mechanisms
In the context of SARS-CoV-2 infection, the immune response defense against the virus survival is a factor of host’s general health condition and the genetic background. Such factors can strengthen specified antiviral immunity during the viral incubation period and in non-severe stages. When the protective immunity is weakened, the virus fastens its replication and spread resulting in a tremendous destruction of the infected tissues especially lungs, kidneys and intestine owing to the increased expression of ACE2 in these organs. 89 However, during the severe infection stage, the virus protects its identity through escaping host’s immune system and inhibiting its components.90,91 The nCoV escapes the innate immunity through suppression of TNF receptor-associated factors (TRAF 3/ 6), which is responsible for activation of both NFκB and IRF-3/7. NFκB is triggered either through TLR3/7 bonding or cytokine receptor signaling, while IRF-3/7is stimulated via RNA sensors (RIG-I and MDA-5) or TLR3/7 ligation. 92 Highly pathogenic HCoVs generally encode viral proteins capable of disturbing RNA sensors, thus antagonizing RNA induced T1IFN synthesis. 93 Additionally both alphacoronaviruses and betacoronaviruses possess capability to degrade host mRNA transcripts leading to deactivation of host interferon response and suppression of T1IFN induction. 94 Other than affecting T1IFN induction, SARS-CoV-2 also deactivates phosphorylation of STAT (signal transducers and activators of transcription) family transcription factors resulting in opposing T1IFN. 95 Moreover, SARS-CoV-2 may evade the adaptive immune response by inducing TNF-mediated T-cell apoptosis. 96
Consequences of SARS-CoV-2 immunopathology
In order to extend its replication and survival period, SARS-CoV-2 has developed a mechanism to evade innate antiviral response of the human host cell. Consequently, this stimulates innate inflammatory responses via recognition of PRRs of the immune cells, resulting in a surge of proinflammatory cytokines/chemokine called cytokine storm (CS). One of the most common complications of CS is pulmonary failure and acute respiratory distress syndrome, acute cardiac injury, vascular disorders, and severe organ damage.
Cytokine storm (CS)
Once SARS-CoV-2 evaded the innate immunity and controlled cell machinery for synthesis of virion particles, at later stages the viruses destroy the cell causing cellular death, release of intracellular content, and liberation of viral molecules, thus facilitating their dissemination. 97 The enormous cytopathies and cellular debris trigger inflammasomes, leading to activation of macrophage derived eicosanoids. 98 Eicosanoids are bioactive lipid mediators (e.g. leukotrienes and prostaglandins) that fire the innate immune response and release pro-inflammatory cytokines inducing hyper-cytokinemia.99–101 Such nonspecific immune response is known as “cytokine storm,” and is characterized by massive release of 150 inflammatory cytokines and other chemical mediators, it is marked by highly proliferating and activated T-cells, NK cells and macrophages102,103 (Figure 3). Despite CS is the final path through which the host immune system can fight against the virus, it is also considered fatal and accounts for the higher morbidity and mortality rates of SARS-CoV-2. If left without treatment, CS would result in severe pathological complications such as sepsis, shock, huge tissue damage, that may progress to organ failure, general edema, and finally death. 103
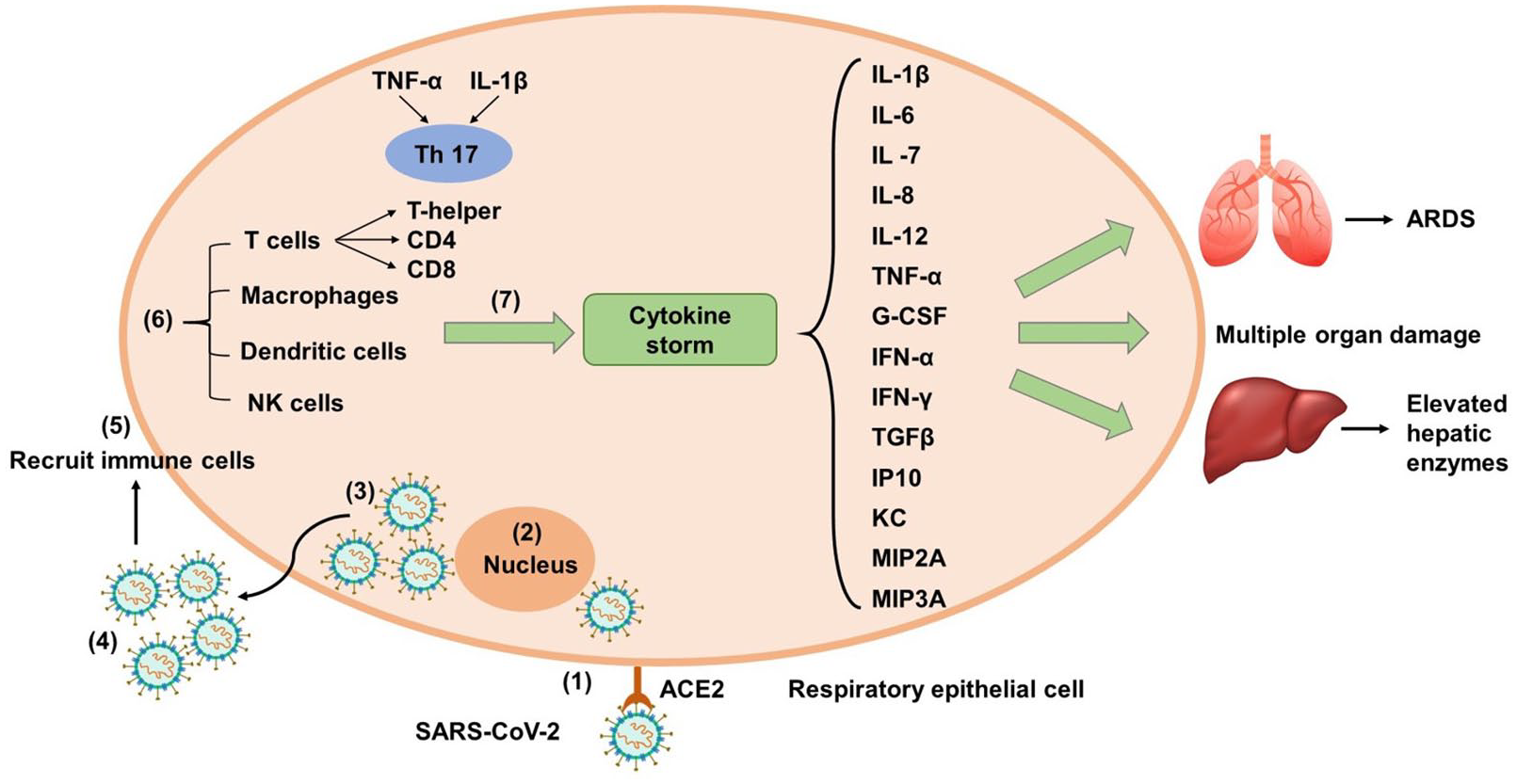
Mechanism of SARS-CoV-2 induced cytokine storm. (1) SARS-CoV-2 evades host immune response. (2) It controls cell machinery and induces synthesis of virion particles. (3) New viral particles destroy the cell causing cellular death. (4) Release of intracellular content and liberation of viral molecules. (5) Triggering of inflammasomes and hyperinflammation. (6) Stimulate host immune response. (7) Release of inflammatory cytokines.
CS hyperinflammation in COVID-19 patients is distinguishable from other types of viral induced CS, in which the increase in ferritin levels is moderate and a massive life threatening organ damage is centralized in the lungs. 104 Because of hyperinflammation, patients develop lymphopenia, thrombocytopenia, coagulopathy, manifested as low fibrinogen and platelet levels, and high D-dimer levels. Additionally, liver tissue damage may occur resulting in elevated levels of hepatic enzymes (aspartate aminotransferase, alanine aminotransferase, and lactate dehydrogenase), and increased ferritin levels secondary to macrophage/hepatocyte activation.105,106 While during the early stages of inflammation, SARS-CoV-2 infected cells showed weakened interferon production facilitating the disease progression. 107
The release of cytokines is closely linked to Th17 responses. 104 Both TNF-α and IL-1β activates Th17 to increase vascular permeability. Activated Th17 cells produce different types of IL for example, IL-17, IL-21, IL-22, and granulocyte-macrophage colony-stimulating factor (GM-CSF). The proinflammatory response of IL-17 triggers the release of inflammatory cytokines such as IL-1β, IL-6, TNF-α, G-CSF, 108 in addition to other pro-inflammatory cytokines (e.g. IL-12, IFN-γ, and transforming growth factor beta (TGFβ)). Along with the release of chemokines for example, IL-8, IP10, keratinocyte chemoattractant (KC), macrophage inflammatory protein (MIP) 2A and MIP3A, attracting other immune cells resulting in tissue damage108–110 The alteration in cytokines and chemokines levels is significant in severe cases of SARS-CoV-2 infected patients when compared to moderate cases. Patients with severe lethal conditions also showed increased levels of IFN (e.g. IFN-α and IFN-γ) and IFN-stimulated genes (ISGs) (such as CXCL10 and CCL-2) upon comparison with mild/moderate diseased patients or healthy individuals.111–113 Moreover, TNF-α and IL-7 induce the release of serum amyloid, fibrinogen, mucins, and antiapoptotic proteins 114 (Figure 3).
Subsequently, these findings suggest that the immunopathogenesis of SARS infection in humans is related to the dysregulation in inflammatory response inside our bodies. Also, the irregular and/or excessive cytokine and chemokine responses by SARS-CoV-2 infected cells are the key player in its pathogenesis. Based on these facts, targeting different key steps in the cytokine storm cascade will potentially improve outcomes in SARS-CoV-2 patients. 115
Acute respiratory distress syndrome (ARDS)
The nonspecific innate immune response stimulation secondary to viral massive tissue damage results in acute respiratory distress syndrome. A severe respiratory failure characterized by alveolar damage, which occurs as the result of immediate diffused inflammation in the lungs and can finally leads to death.60,78 The main symptoms of ARDS are short, rapid breathing, and cyanosis. Patients expressing such complications are referred to the intensive care units for mechanical ventilation. While in severe cases where patients are unable to breath and need life support assistance, are usually connected to extracorporeal membrane oxygenation (ECMO) machine.89,116 Histopathological examination of pulmonary lesions in SARS patients showed nonspecific inflammatory reactions as pulmonary edema, inflammatory cell infiltration and hyaline membrane formation. Additionally, alveolar damages were recorded including alveolar epithelial cells exfoliation, widening/damage to the alveolar septa, and infiltration in the alveolar space. 60 Therefore, pathological modifications caused by inflammatory responses as degeneration (necrosis), infiltration and hyperplasia provided an explanation why some patients are manifested with severe lung injury although their viral loads were low. 109
Computed tomography (CT) scan on chests of SARS infected patients showed characteristic white patches filled with fluids called “ground glass.” 35 A recent study confirmed this through autopsies, reporting the presence of clear liquid gelatinous material filling the lungs, closely similar to lungs of wet drowning. 78 Although the composition of such clear liquid jelly has not been yet identified, previous researches explained the strong correlation between hyaluronan (HA) and ARDS. 117 HA is a glycosaminoglycan ingredient present at high concentrations in the extracellular matrix of the lungs and it is capable of absorbing amounts of water 1000 times of its molecular weight. The production and organization of HA is disturbed during SARS infection, moreover the elevated levels of proinflammatory cytokines during cytokine storm stimulate HA-synthase-2 (HAS2) in lung alveolar epithelial cells, CD31+ endothelium and fibroblasts. 118 Subsequently, the use of medical grade hyaluronidase could decrease the accumulation of HA in the lungs, thus reduce one of the pulmonary complications in SARS-CoV-2 patients. 89
Coagulation and vascular disorders
One of the prevalent characteristics of CS is the cardiovascular disorder, in which the patients are manifested with disseminated intravascular coagulation, capillary leakage, and reduction in blood pressure. 119 Generally, the immune-mediated pathways associated with systemic infections may result in cytolytic events such as endothelial cell death. Once they are exposed to the circulating cytokines, body systems maintain their silence and develop a conserved hemostatic response. This hemostatic mechanism involves platelets recruitment and fibrin deposition to achieve blood coagulation and reduce blood loss. 120 The surge of systemic cytokines is usually accompanied by decrease in platelet numbers expressing their consumption. Although these events are considered host-protective mechanisms, prolonged systemic infection may result in extensive thrombus formation followed by vascular closure, disseminated intravascular coagulation, tissue damage, and finally host’s death. The vascular endothelial cells of the lungs and kidneys were found to be the most vulnerable areas to the complement mediated injury, and the highly affected endothelial dysfunctions. 121
Infection with COVID-19 increases the risk of hypercoagulation and the incidence rates of arterial and venous thrombosis, especially in younger patients. 122 Several coagulation disorders were manifested in COVID-19 patients, such as ischemic stroke in large vessels that was reported in patients with mild symptoms or even in asymptomatic patients. 123 Also, advanced stages of COVID-19 infection were accompanied by higher proportions of acute venous thromboembolism. 124 A recent study reported that 25% of patients admitted to the intensive care unit (ICU) who haven’t receive any anticoagulant prophylactic therapy, developed deep vein thrombosis in the lower extremities, while 40% of the patients experienced death. 125 The most widespread thrombotic complication following COVID-19 infection is the pulmonary embolism, which may result in acute right ventricular failure. 126 However, some patients may suffer also form ischemic stroke, myocardial infarction, deep vein thrombosis, and systemic arterial embolism. 127 Despite no previous history of heart failure, 25% of COVID-19 hospitalized patients, and 33% of patients at the ICU were diagnosed with heart failure.128,129 A possible explanation could be attributed to SARS-CoV-2 direct effect or due to systemic inflammation that affects the heart. Consequently, severe acute myocarditis, and cardiogenic shock may occur, resulting in multi-organ damage and finally death.130,131
Suggested therapies and treatments
The urge for the development of an effective therapy against COVID-19 infection is focused on understanding coronavirus pharmacological and toxicological mechanisms in human body. 5 Drug repurposing is one of the widely utilized approaches, to redesign the use of previously approved drugs in COVID-19 treatment, based on information of their mechanism of actions.132–136 An example of these repurposed drugs (summarized in Table 1) is relying on the mechanism of viral internalization by endocytosis, 137 suggesting repurposing of previously approved drug for blocking SARS-CoV-2 endocytosis. Another examples include the use of antidepressants such as fluvoxamine, sertraline, and imipramine,137–140 and antifungals such as nystatin, and itraconazole as promising options in COVID-19 treatment. In addition to other classes of medications for example, antihistamines such as promethazine and terfenadine,137,140–142 diuretics such as amiloride, microtubule formation inhibitor such as vinblastine, antihelminthic such as flubendazole, and beta-methyl cyclodextrin as a bioavailability enhancer were all found to have high efficacy in improving therapeutic outcomes in COVID-19 patients.137,140
Different ongoing clinical trials were conducted for the approval of COVID-19 treatment.

Despite the ongoing clinical trials, no therapy was found successful against SARS-CoV-2 infection up to date.143,144 There is no strong evidence that a potential or prophylactic therapy is available to enhance outcomes in patients with suspected or confirmed COVID-19. However, the rapidly growing awareness about SARS-CoV-2 virology may provide multiple potential targets for medicines. Remdesivir is the most effective treatment discovered so far as it has proven a potent effect in vitro, and currently it is only being evaluated in ongoing randomized trials, yet it has been licensed recently by the United States Food and Drug Administration (FDA). 145 Oseltamivir has not been proven to be effective and officially corticosteroids are not approved. 143 Current research suggests the use of TMPRSS2 inhibitors such as camostat mesylate and nafamostat mesylate, angiotensin-converting enzyme inhibitors or angiotensin receptor blockers, inhibitors of replication, membrane fusion and assembly of SARS-CoV-2 such as remdesivir and umnifenovir, and 3-Chymotrypsin like protease (3CLpro) inhibitors in COVID-19 patients. 146 Currently various clinical trials are conducted to test the potential treatments of COVID-19, which are summarized in Table 1.143,146 However, the new treatment guidelines for COVID-19, according to the National Institutes of Health (NIH), are focused on the following treatment options:
Antithrombotic therapy
Infection with COVID-19 has been associated with inflammation and a prothrombotic state. 145 Although the occurrence of thrombotic complications is unclear, some reports have been seen of COVID-19 related thromboembolic disease in patients admitted to the ICU. 147 Several clinical trials are being conducted nowadays, some are recruiting subjects as in St. Michael’s Hospital, Geneva Hospitals, and NYU Langone Health for example, (“Coagulopathy of COVID-19: A Pragmatic Randomized Controlled Trial of Therapeutic Anticoagulation Versus Standard Care—Full Text View—ClinicalTrials.gov,” 2020; “Preventing COVID-19 Complications With Low- and High-dose Anticoagulation—Full Text View—ClinicalTrials.gov,” 2020; “A Randomized Trial of Anticoagulation Strategies in COVID-19—Full Text View—ClinicalTrials.gov,” 2020). Other trials are still in progress as conducted by (“Intermediate or Prophylactic-Dose Anticoagulation for Venous or Arterial Thromboembolism in Severe COVID-19—Full Text View—ClinicalTrials.gov,” 2020). The American Society of Hematology suggested the use of low molecular weight heparin or any alternative as a prophylactic therapy against thrombosis for all COVD-19 hospitalized patients, after comparing the coagulation complications to the high risk of bleeding in some cases. 148
Antiviral drugs
Based on the recommendations of the National Institutes of Health, 145 Remdesivir is available as FDA (food and drug administration) emergency drug for treatment of COVID-19 patients with severe cases. On the other hand, Chloroquine/Hydroxychloroquine are used with some restriction (only low doses) to avoid toxicities.
Immune-based therapy
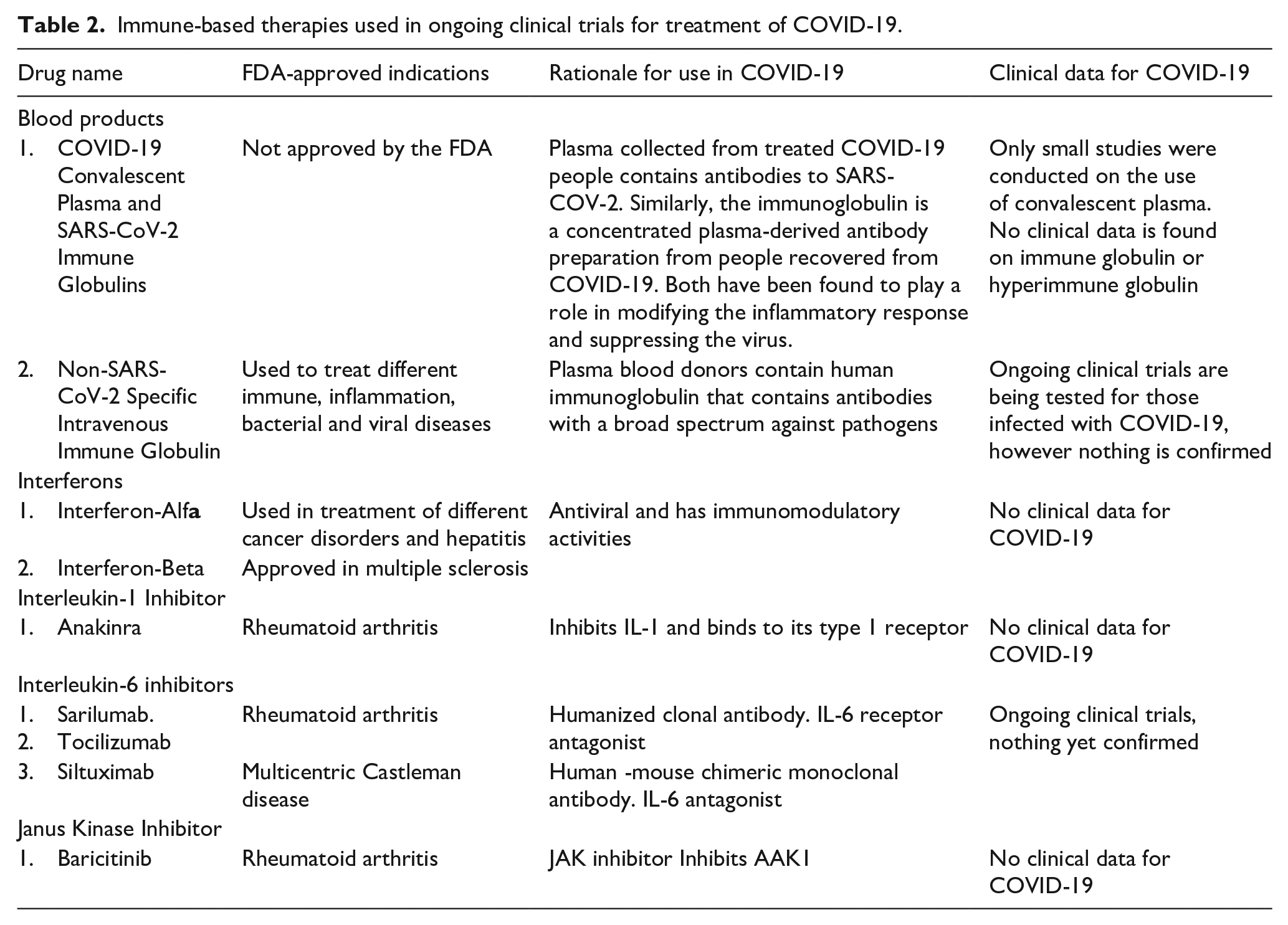
According to NIH guidelines 149 summarized in Table 2, no reliable data is available to use the convalescent plasma or intravenous immune globulins of cured COVID-19 in the treatment. Although some research studies were conducted on patients showed a promising effect, yet the efficacy and safety of convalescent plasma need further investigation.150–152 Cytokine targeting therapies (Table 2) are based on blocking IL-1, IL-6, and TNF-α, as these molecules are highly upregulated in patients with COVID-19. Although different clinical trials are ongoing to assess the effectiveness of these therapies. One example of IL-1 receptor antagonist is anakinra, which showed a promising effect in blocking the biologic activity of IL-1.153–156 Examples of IL-6 inhibitors are tocilizumab, siltuximab, and sarliumab, which also had a potential therapeutic effect in COVID-19 patients.157–159 Some ongoing trials are being tested for the effectiveness of TNF-α blockers. 160 Other immune based therapies used for treatment of SARS-CoV-2 infected patients are Janus kinase inhibitors such as barcitinib. 161 Furthermore, mesenchymal stem cells162,163 possessed potent immunomodulatory activities, intravenous immunoglobulin and convalescent plasma therapies were other therapeutic options shown effectiveness in COVID-19 patients.164–167
Immune-based therapies used in ongoing clinical trials for treatment of COVID-19.
Complementary and alternative medicine
Recently scientists are proposing the use of complementary and alternative medications as an adjunct therapy for COVID-19. Many countries relied on the use of natural preparations such as amla, tulsi, coronil, and giloy. 168 While some studies addressed the use of vitamin C, zinc, and immunomodulatory supplements to increase patient’s quality of life and reduce the complications. 169
Conclusion and future prespectives
The outbreak of SARS-CoV-2 has raised an alert on the importance of studying viral transmission across the species. The virulence of SARS-CoV-2 relies on its ability to infect the lower respiratory tract which may progress to pneumonia and severe lung injury. Binding of viral S protein to ACE2 receptor, expressed on the cellular surfaces of multiple organs, results in severe organ damage in its advanced stages. The interaction between SRAS-CoV-2 and the host antiviral immune response is essential for the pathogenicity. The novelty of SARS-CoV-2 to human beings resulted in absence of acquired immunity against it and to more complex and ambiguous perspective to the host immune responses. Whereas, with immunocompromised patients and in severe cases, SARS-CoV-2 possesses an ability to evade the host immune responses followed by several inflammatory complications progressing to CS. The severity of CS is a factor of patient’s age, genetic variability and the presence of other pathological conditions or comorbidities.
COVID-19 disease affected all walks of life globally.222–227 Currently, there has not been a single approved treatment proved to be effective against SARS-CoV-2 infection. However, according to the World Health Organization, social distance, and hand washing are the main protective measures against COVID-19. Improving the ventilation rate, avoiding air recirculation, reducing the number of people and the direct contact with them will aid in reducing the disease transmission. Most available treatment protocols depend on the use of combination therapies including antiviral, anti-inflammatory, and immunomodulatory based on the available limited clinical trials and comparative analyzes with SARS-CoV. It is recommended to maintain the balance between the use of anti-inflammatories and sustaining the effective antiviral immune response. Targeting specific key steps in the cytokine storm pathway could provide a novel therapeutic option to decrease patients’ suffering and improve treatment outcomes. Moreover, studying MHC class I and II alleles in SARS-CoV-2 and determining HLA loci would provide an insight for development of anti-SARS-CoV-2 vaccines and designing effective clinical management plan against COVID-19.
Footnotes
Acknowledgements
The authors want to thank their respective institutions for their continued support. The language and technical editing support provided by The Editing Refinery, MD, USA is highly acknowledged.
Authors’ contributions
AD and NY performed literature research, gathered, and analyzed information, and generated short preliminary write-ups. MS, MME, and MWQ conceptual work, framework, draft write-up, critical reading, and editing. All authors read and approved the final manuscript.
Availability of data and materials
This is a review article. All data generated or analyzed during this study are included in this published article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.