
Abstract
The COVID-19 pandemic, caused by the novel coronavirus SARS-CoV-2, has posed unprecedented challenges to global health. Understanding the complex molecular and cellular mechanisms underlying SARS-CoV-2 infection is critical for developing effective therapeutic strategies. This narrative review comprehensively examines the key pathogenesis pathways involved in SARS-CoV-2 infection, with a focus on viral entry mechanisms, innate and adaptive immune responses, and the dysregulated inflammatory cascades that drive disease severity. We highlight the pivotal roles of immune cells, such as neutrophils, macrophages, natural killer cells, and T lymphocyte subsets, in orchestrating both protective and pathological responses. Central to severe COVID-19 is the excessive release of pro-inflammatory cytokines, known as the cytokine storm, which leads to acute respiratory distress syndrome and multi-organ failure. Furthermore, we critically evaluate conflicting evidence regarding key immunopathological processes. We further explore the impact of viral variants on immune evasion and therapeutic efficacy, as well as the emerging biomarkers that may guide personalized treatment. Finally, we discuss current and prospective therapeutic interventions, analyzing the clinical evidence and limitations of strategies targeting viral replication, immune modulation, and inflammation resolution. This synthesis provides valuable insights into the pathophysiology of COVID-19 and underscores the importance of integrated, adaptive approaches to combat this evolving disease.
1. Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has triggered an unprecedented global pandemic, resulting in over 775 million confirmed cases and more than 7 million deaths worldwide as of early 2024.
1
As a member of the Coronaviridae family, SARS-CoV-2 is an enveloped, positive-sense RNA virus capable of infecting both humans and animals (Figure 1). SARS-CoV-2 variants have continuously evolved through genetic and antigenic changes, enabling immune evasion from prior infection or vaccination.
2
This drives breakthrough infections despite existing immunity.3,4 Notably, Omicron variants exhibit tropism for the upper respiratory tract (e.g., nasal epithelium and oropharynx), contrasting with earlier variants, such as Delta, which predominantly targeted the lower respiratory tract (e.g., alveoli).
5
The structure of the COVID-19 virus.
Despite the passage of time, novel SARS-CoV-2 variants continue to emerge rapidly, such as JN.1, which has gained global prominence. Derived from BA.2.86—the only other strain to accumulate over 30 novel mutations in the spike protein since Omicron’s initial appearance more than two years ago—JN.1 has become the dominant variant in the United States. 6 While later variants like JN.1 demonstrate increased transmissibility, hospitalizations and deaths have remained relatively lower compared to earlier pandemic waves, largely due to accumulated population immunity from vaccination and prior infection. To address the evolving viral landscape and waning vaccine efficacy, vaccine formulations and public health policies have been updated to sustain protection against severe disease and mortality.7,8
Viral RNA (vRNA) has been detected in blood, serum, and plasma samples from infected individuals.9,10 Moreover, SARS-CoV-2 exhibits a preferential infection of cells expressing blood group A antigens.11,12 Approximately 80% of COVID-19 cases are mild or asymptomatic, presenting symptoms similar to the common cold, including dry cough, headache, anosmia, dyspnea, fatigue, and fever. 13 These cases are generally controlled by an effective immune response. However, about 15% of patients develop severe disease requiring intensive care and oxygen therapy, while roughly 5% progress to critical illness characterized by life-threatening pneumonia, acute respiratory distress syndrome (ARDS), and septic shock. 14
The overactivation of these signaling pathways by SARS-CoV-2 results in organ damage, cytokine storm (CS), and ultimately death (Figure 2).
15
Furthermore, the virus’s high mutability and the continuous emergence of new variants present significant challenges for the identification, prevention, and treatment of COVID-19.16,17 The more recent Omicron variants are particularly concerning due to their increased infectivity and enhanced ability to evade the human immune response. SARS-CoV-2-induced cytokine storm via overactivated signaling pathways.
The inflammatory response to SARS-CoV-2 infection involves a coordinated activation of multiple defense mechanisms. Initially, macrophages act as the first line of defense by engulfing pathogens and releasing cytokines that recruit other immune cells.18,19 These cytokines, exhibiting both pro- and anti-inflammatory properties, constitute the second line of defense by stimulating B and T lymphocytes to produce antibodies and induce apoptosis of infected cells, thereby aiding pathogen clearance. The third and most significant mechanism is the eicosanoid pathway, which catalyzes the conversion of arachidonic acid into prostaglandins, prostacyclins, and thromboxane, molecules essential for resolving inflammation. 20
Inflammatory mediators involved in this response are classified into three main groups. Preformed mediators such as histamine, serotonin, and lysosomal enzymes are stored in mast cells, platelets, neutrophils, and macrophages, ready for immediate release even without inflammatory triggers. Newly synthesized mediators, including eicosanoids (prostaglandins, leukotrienes) and cytokines, are produced during inflammation by leukocytes, platelets, endothelial cells, and macrophages. Complement factors, activated via a cascade of plasma proteins, release intermediates like C3a, C5a, and the membrane attack complex C5b-9, which further stimulate complement-clotting-kinin pathways leading to blood clotting and pain.21,22
Severe COVID-19 is characterized by an exaggerated and systemic inflammatory reaction, with elevated cytokine levels detected in the lungs and bloodstream. 23 This cytokine production is triggered by the recognition of viral pathogen-associated molecular patterns (PAMPs) and host-derived danger-associated molecular patterns (DAMPs) signaling tissue injury. 24 SARS-CoV-2 infection induces the release of potent proinflammatory cytokines such as interleukin-6 (IL-6), IL-8, tumor necrosis factor-alpha (TNF-α), interferon-gamma (IFN-γ), and IL-17, which can precipitate sepsis.25,26
While the immune system’s primary role is to defend against pathogens, uncontrolled immune activation can result in a cytokine storm (CS), a hyperinflammatory state that significantly contributes to COVID-19 severity. The activation of inflammatory signaling pathways and the ensuing cytokine storm are pivotal in the development of acute respiratory distress syndrome (ARDS) and multiple organ failure (MOF) in affected patients (Figure 3).27–31 Systemic organ failure as a consequence of SARS-CoV-2 pathogenesis.
Currently, no specific cure for COVID-19 exists, but several therapeutic strategies have been proposed, including cytokine inhibitors, JAK inhibitors, immunomodulators, and plasma therapy.32–34 This narrative review aims to provide a comprehensive understanding of how SARS-CoV-2 induces inflammatory storms and its interplay with pre-existing inflammatory conditions. The primary scope of this review is to critically synthesize the molecular and cellular mechanisms of SARS-CoV-2-induced inflammation and immune dysregulation. We aim to move beyond description by evaluating the strength and consistency of the evidence, discussing competing hypotheses, and providing an in-depth analysis of the promises and pitfalls of current therapeutic strategies.
2. Methods
This study is a narrative review. To ensure a comprehensive and systematic literature search, we queried major electronic databases, including PubMed/MEDLINE, Scopus, and Web of Science, for articles published so far. Key search terms included: “SARS-CoV-2,” “COVID-19,” “pathogenesis,” “immune response,” “inflammation,” “cytokine storm,” “T cells,” “macrophages,” “neutrophils,” “therapy,” “treatment,” and “variants.” The search strategy combined these terms using Boolean operators (AND, OR). Included studies were original research articles, clinical trials, and meta-analyses published in English. Review articles and commentaries were consulted for background context but were not the primary source of evidence. The selection process prioritized high-impact studies that elucidated key mechanisms or provided robust clinical data, with particular attention to contradictory findings to facilitate a critical analysis.
3. Viral pathogenesis and immune evasion
3.1. Viral entry and initial infection
SARS-CoV-2 enters host cells primarily via the angiotensin-converting enzyme 2 (ACE2) receptor, with entry facilitated by host proteases like TMPRSS2 or cathepsins. 5 This interaction not only mediates viral entry but also disrupts the renin-angiotensin system (RAS), promoting inflammation and vascular dysfunction. 29
3.2. Immune evasion and the impact of variants
The continuous evolution of SARS-CoV-2 has led to the emergence of variants with enhanced immune evasion capabilities. These variants accumulate mutations, particularly in the spike protein, that can reduce neutralizing antibody binding from prior infection or vaccination, increase transmissibility, and alter cell tropism.2,3 For instance, while the Omicron variant (BA.1 and its sublineages) demonstrated significant escape from humoral immunity, its relative attenuation in the lower respiratory tract may be linked to altered cellular tropism and replication efficiency.5,6 This evolution directly impacts therapeutic efficacy; for example, some monoclonal antibody therapies lost potency against later variants like Omicron, necessitating the continuous development of updated countermeasures.7,8 Understanding these escape mechanisms is crucial for designing next-generation vaccines and antivirals that target more conserved viral regions.
4. Inflammation signaling pathways
Inflammation manifests through symptoms such as fever, cardiovascular complications, allergies, anaphylaxis, fibrosis, and autoimmune diseases. 35 This complex process can be divided into four stages: initiation by triggers like infections or tissue damage; activation of sensors, including mast cells and macrophages; release of pro-inflammatory substances such as cytokines and chemokines; and the resultant effects on affected tissues. The nature of the inflammatory response varies depending on the pathogen involved; for example, bacterial infections primarily activate toll-like receptors (TLRs), whereas viral infections stimulate type I interferons (IFNs). 36 Notably, chronic inflammatory conditions of unknown etiology are increasingly prevalent and often coexist with diseases such as obesity, type 2 diabetes, atherosclerosis, neurodegenerative disorders, and cancer. 37
Therapeutic strategies targeting later disease stages focus on mitigating excessive host responses, including cytokine overproduction, systemic inflammation, dysregulated platelet aggregation, and coagulopathy. Efforts also aim to modulate inflammatory markers and cytokine expression, underscoring the importance of distinguishing between pro- and anti-inflammatory responses to develop effective treatments.38,39
Key inflammatory pathways involved in COVID-19 pathogenesis (Figure 4) include: 1. Nuclear Factor kappa B (NF-κB) Pathway: A pivotal transcription factor regulating genes involved in inflammation. SARS-CoV-2 activates NF-κB/Aquaporin1 (Aq1), promoting the production of pro-inflammatory cytokines and chemokines. Dysregulated NF-κB signaling can lead to cytokine storm and severe lung injury.
40
2. Mitogen-Activated Protein Kinase (MAPK) Pathway: Transmits extracellular signals to the nucleus, regulating gene expression and enhancing pro-inflammatory mediator production.
41
3. Janus Kinase-Signal Transducer and Activator of Transcription (JAK-STAT) Pathway: Mediates immune activating factors (IAF), cytokine and growth factor signaling, activating transcription factors critical for immune responses and inflammation.
42
4. Cyclooxygenase-2 (COX-2) Pathway: Catalyzes the conversion of arachidonic acid—released by phospholipase A2 (PLA2) from membrane phospholipids—into prostaglandins (PGE2/PG12), thromboxanes, and leukotrienes, which regulate immune homeostasis and inflammation.
43
5. NOD-like Receptor Protein 3 (NLRP3) Inflammasome Pathway: Detects danger signals and activates production of pro-inflammatory cytokines IL-1β and IL-18, amplifying inflammatory responses.
44
6. Angiotensin-Converting Enzyme 2 (ACE2) Pathway: SARS-CoV-2 exploits ACE2 to enter cells; this interaction disrupts the renin-angiotensin system, promoting inflammation.
45
7. Toll-Like Receptor (TLR) Pathway: Recognizes viral components, initiating immune responses by inducing inflammatory cytokine production and immune cell recruitment.
46
8. Interferon (IFN) Pathway: Central to antiviral defense, interferons activate immune cells and inhibit viral replication. A notable feature of severe COVID-19 is a delayed or impaired type I IFN response, which may allow uncontrolled viral replication early in infection, followed by a pathologic influx of inflammatory cytokines later.47,48 Primary mechanisms of pathogenesis and inflammatory cascades in SARS-CoV-2 infection.
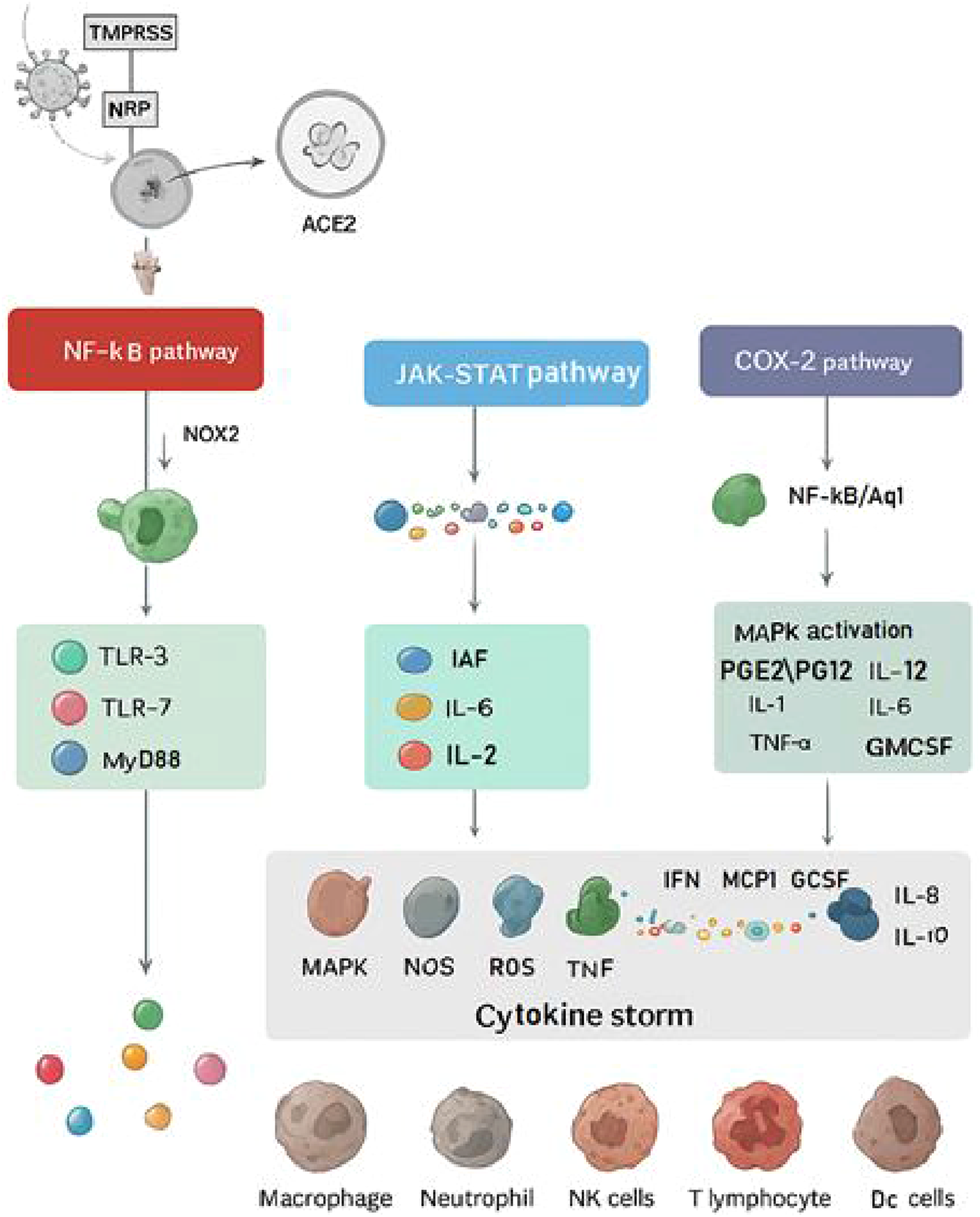
These interconnected pathways orchestrate the inflammatory response; whose dysregulation underlies many COVID-19 complications.
Three primary transcription factors play crucial roles in the inflammatory response pathway: NF-κB, JAK-STAT, and hypoxia-inducible factor-1 alpha (HIF-1α). 49 While NF-κB predominantly regulates the acute phase of inflammation aimed at restoring homeostasis, its sustained activation, along with JAK-STAT and HIF-1α, can contribute to chronic inflammation and diseases such as long COVID-19. NF-κB promotes the expression of key inflammatory cytokines, including IL-1, IL-6, TNF-α, lymphotoxin, and IFN-γ. Notably, IL-1 and TNF-α engage in a positive feedback loop by stimulating NF-κB activation, amplifying the inflammatory response. 50
HIF-1α is activated under hypoxic conditions, a common feature within inflamed tissues where immune cells experience reduced oxygen availability. This transcription factor induces genes involved in angiogenesis and erythropoiesis, thereby enhancing immune cell survival in hypoxic environments. 51 Cytokines such as IL-1β further support HIF-1α transcriptional activity, with NF-κB facilitating this process by binding to HIF-1α promoters under inflammatory stimuli. 52
The STAT protein family mediates diverse roles depending on the inflammatory trigger. Specific cytokines activate distinct STAT proteins, which can either promote or suppress inflammation. For example, viral infections induce interferons that activate STAT1 and STAT2, enhancing antiviral defenses and modulating inflammation. Conversely, STAT6 influences T helper cell differentiation, which can either exacerbate allergic inflammation or mitigate autoimmunity depending on the subset involved.53–55
Key pathogenic mechanisms and inflammatory pathways in SARS-CoV-2 infection.

5. Induction of inflammation by arachidonic metabolites
Arachidonic acid is released from membrane phospholipids through the action of phospholipases A2 and undergoes a series of metabolic transformations. 61 The production of prostaglandins and eicosanoids is mediated by enzymes such as COX and lipoxygenases (LOX). COX exists in two isoforms: COX-1, constitutively expressed in most cells to maintain physiological functions, and COX-2, which is inducible by inflammatory stimuli, hormones, and growth factors, and serves as the main source of prostanoids during inflammation and proliferative diseases. 20 Non-steroidal anti-inflammatory drugs (NSAIDs) like aspirin, ibuprofen, and indomethacin non-selectively inhibit both COX-1 and COX-2, whereas COXIBs such as celecoxib and valdecoxib selectively target COX-2. 62 Studies have reported elevated expression of COX isoforms in severe SARS-CoV-2 infection, with a notable overexpression of 15-LOX-1 in male patients with severe disease and female patients with non-severe infection. Moreover, 15-LOX-2 overexpression was significant only in males with severe COVID-19. 63 While COX-1 contributes to tissue integrity, cell proliferation, and vasodilation, COX-2 is primarily involved in inflammatory responses triggered by cytokines and growth factors. 64 Prostaglandins and cytokines synergistically activate NF-κB, inducing the expression of inflammation-related genes in immune cells such as macrophages and neutrophils. 65 Given the pivotal role of the eicosanoid and prostaglandin pathways in modulating inflammation and facilitating viral replication, a deeper understanding of their expression patterns and mechanisms during COVID-19 could inform the development of targeted therapies and preventive strategies, shedding light on the molecular basis of disease progression.
6. The cytokine storm and dysregulated immunity
The term “cytokine storm” itself is debated, with some studies arguing it may be an oversimplification of a more complex immunodynamic dysfunction. The overproduction of pro-inflammatory cytokines and chemokines disrupts immune homeostasis, promoting infiltration of inflammatory cells into lung tissue and impairing immune function. Additionally, dysregulation of the RAS, caused by ACE2 downregulation, correlates with increased mortality in COVID-19. 29 Both mechanisms converge to exacerbate hyperinflammation, leading to increased vascular permeability, edema, hypercoagulation, and multi-organ damage.30,31
6.1. Regulatory T cells in COVID-19
Under physiological conditions, regulatory T cells (Tregs) migrate to inflamed tissues to suppress excessive inflammation and promote tissue repair. 66 Tregs inhibit the activity of CD4+ and CD8+ T cells and reduce the presence of NK cells, eosinophils, and neutrophils, thereby controlling immune responses to viral infections. However, COVID-19 patients exhibit a marked reduction in circulating Tregs, 67 which impairs inflammation control, disrupts the Treg/Th17 balance, and increases the risk of respiratory failure. Tregs are classified into three subsets based on Foxp3 and CD45RA expression: resting Tregs (rTregs), activated Tregs (aTregs), and cytokine-secreting non-suppressive T cells (nonTregs). Among these, aTregs possess the highest suppressive capacity, while non-Tregs secrete cytokines but lack inhibitory function. 68 Both moderate and severe COVID-19 cases show decreased levels of CD4+ CD25+ CD127^low Tregs and CD45RA+ Tregs, with severe cases presenting a significantly lower proportion of CD45RA+ Tregs compared to moderate cases; CD45RO+ Treg levels remain comparable between groups.69,70 The reduction of CD45RA+ Tregs alongside elevated IL-10 may exacerbate immune dysregulation and increase mortality risk in severe COVID-19. 71 In asymptomatic SARS-CoV-2 infection, a rise in aTregs may restrain early T cell activation, potentially limiting immune overreaction. 72 Elevated IL-6 disrupts the Treg/Th17 ratio, contributing to complications such as diabetes and immune dysfunction. Additionally, SARS-CoV-2 directly targets lymphocytes, further impairing immune regulation by suppressing Foxp3 expression and promoting CD4+ T cell overactivation. 73 Multiple factors contribute to Treg depletion in COVID-19, including lymphocyte apoptosis from excessive inflammation, Treg destabilization under inflammatory conditions, IL-6-mediated inhibition of Treg expression, SARS-CoV-2-induced CD4+ T cell activation reducing Foxp3 levels, direct viral lymphocyte attack, and hypoxia-induced inhibition of Treg differentiation in the lungs.74–78 Therapeutically, Tregs hold promise in mitigating pathological inflammation, cytokine storm, fibrosis, and platelet activation in COVID-19. 79
6.2. Th17 cells in COVID-19
Th17 cells, a subset of immune cells, play a dual role in immunity, acting as both protectors and contributors to pathology. They are implicated in chronic inflammation and autoimmune diseases through the production of cytokines such as IL-17A, IL-17F, IL-21, IL-22, GM-CSF, IL-10, and IFN-γ.
80
Among these, IL-17 is a potent pro-inflammatory cytokine that drives widespread inflammation by inducing the release of granulocyte colony-stimulating factor (G-CSF), which promotes neutrophil production and mobilization. IL-17 also stimulates the secretion of pro-inflammatory mediators, including IL-1β, IL-6, and TNF-α, contributing to systemic symptoms like fever. Additionally, IL-17 enhances chemokine production (KC, MIP-2α, IL-8, IP-10, MIP-3α), recruiting further immune cells to sites of infection, and activates matrix metalloproteinases, leading to tissue damage and remodeling. Both IL-17 and GM-CSF are involved in autoimmune and inflammatory pathologies.
81
IL-17 attracts neutrophils and monocytes to infected tissues and promotes cytokines such as G-CSF and IL-6 that sustain innate inflammation. Chemokines, including CXCL1, CXCL2, and CXCL10, also facilitate myeloid cell recruitment. While CD4+, CD8+, and innate lymphoid cells (ILCs) are primary IL-17 sources, neutrophils can produce IL-17 under certain conditions.
82
Emerging evidence suggests that Th17-mediated inflammation significantly contributes to COVID-19 pneumonia progression. The release of IL-17 and GM-CSF exacerbates neutrophil migration and suppresses regulatory T cell (Treg) responses. In contrast, Tregs express anti-inflammatory cytokines such as IL-4, IL-10, and TGF-β, playing a critical role in controlling excessive immune activation.
83
Severe COVID-19 is characterized by reduced Treg numbers and a decreased Treg/Th17 ratio, indicating impaired regulation of pro-inflammatory responses. This shift toward a Th17-dominant phenotype may drive excessive cytokine and chemokine release, amplifying inflammation and tissue injury (Figure 5). The roles of macrophages and T cells in severe respiratory syndrome caused by COVID-19.
Roles of immune cells in COVID-19 pathogenesis.

6.3. CD8+ T cells in COVID-19
CD8+ cytotoxic T cells play a dual role in COVID-19. While their rapid activation is essential for viral clearance and protection against severe disease, hyperactivation of CD8+ T cells has been implicated in disease progression and tissue damage. However, the extent to which CD8+ T cells are drivers of immunopathology versus essential antiviral defenders remains a subject of debate. Critically ill patients exhibit increased numbers of hyperactive CD38^hi CD8+ T cells, which correlate with cytokine storm and damage to organs such as the heart, kidneys, and liver.99,100 Post-mortem analyses reveal the presence of highly cytotoxic CD8+ T cells in affected organs, suggesting their potential contribution to fatal tissue injury, although a direct causal link remains to be definitively established.101–103 Nienhold et al. (2020) described two distinct patterns in deceased COVID-19 patients: one with high viral loads, minimal lung damage, and elevated interferon-stimulated genes (ISGs) and cytokines (Pattern I); the other with low viral loads, significant lung injury, low ISG expression, and increased activated CD8+ T cell infiltration (Pattern II). The latter pattern suggests that CD8+ T cell accumulation may contribute to alveolar damage and pneumonia symptoms over time. 104 This supports a hypothesis where inadequate early innate control leads to a pathologic, T-cell-driven phase of disease. Nonetheless, many studies indicate that innate immune cell infiltration, rather than CD8+ T cell activity, is the primary driver of tissue damage in severe SARS-CoV-2 infection. For example, increased CD8+ T cell counts observed in ARDS patients were associated with bacterial pneumonia rather than SARS-CoV-2 infection. 105
6.4. Macrophages (MQ) in COVID-19
Macrophages are a heterogeneous population of innate immune cells residing in various tissues, including brain microglia, liver Kupffer cells, and lung alveolar and interstitial macrophages, where they maintain tissue homeostasis and defend against pathogens. 87 In COVID-19, pulmonary macrophages derived from infiltrating inflammatory monocytes become hyperactivated, initiating a deleterious cycle of pro-inflammatory cytokine release and recruitment of cytotoxic effector cells, which exacerbates tissue damage at the infection site. 88
This hyperinflammatory state can culminate in macrophage activation syndrome (MAS) (Figure 6), a systemic inflammatory condition also observed in malignancies and pediatric rheumatologic diseases such as systemic juvenile idiopathic arthritis (SJIA).
89
Inflammation disrupts the balance between coagulation and fibrinolysis; cytokines like TNF-α and IL-1 induce tissue factor expression by monocytes and macrophages, contributing to coagulopathy.
90
In the COVID-19 patients, an accumulation of inflammatory monocytes characterized by high expression of HLA-DR and CD11c is noted, especially in mild cases. Recognition of viral and host molecules by pattern recognition receptors triggers a cytokine storm involving IL-6, IL-8, TNF-α, IFN-γ, and IL-17, with cytokine levels correlating with disease severity and immune dysregulation in critically ill patients.
91
Macrophage-mediated cytokine response contributing to severe respiratory complications in COVID-19.
6.5. Polymorphonuclear neutrophils (PMN) in COVID-19
Neutrophils, key players in innate immunity, release neutrophil extracellular traps (NETs) that contribute to inflammation, cell death, and microthrombus formation. In severe COVID-19, activated macrophages can engulf erythrocytes through hemophagocytosis, further amplifying inflammation. 92 The excessive immune response involves NET release, hemophagocytosis, and activation of monocytes and macrophages, which together contribute to both antiviral defense and COVID-19-associated pathology. Additionally, COVID-19 patients exhibit increased neutrophil infiltration and hyperactivation of circulating natural killer (NK) cells.93,94
6.6. Natural killer (NK) cells in COVID-19
Natural killer (NK) cells are key players in antiviral defense and tumor surveillance, but they also possess important immunomodulatory functions. Towards the resolution of infection, NK cells help protect tissues by eliminating overactivated immune cells such as CD8+ T cells and macrophages. SARS-CoV-2 infection commonly induces lymphopenia, characterized by a reduction in circulating lymphocytes, including NK cells, accompanied by increased neutrophils and decreased monocytes, especially in severe COVID-19 cases.95,96 Severe disease is marked by diminished levels of CD4+ Th1 cells, regulatory T cells (Tregs), CD8+ T cells, and NK cells, with the latter’s reduction correlating with disease severity and acute phase. 97 Post-recovery studies show that NK cell counts typically normalize; however, in long-COVID patients, elevated NK cell levels have been reported compared to fully recovered individuals. SARS-CoV-2 induces NK cell exhaustion by upregulating inhibitory receptors and downregulating activating receptors, impairing interferon (IFN) signaling and reducing their antiviral and immunomodulatory efficacy. 98
7. Therapeutic perspectives
Understanding the complex pathways involved in the inflammatory response to SARS-CoV-2 infection is essential for developing targeted therapies aimed at mitigating the detrimental effects of excessive inflammation. This review underscores the pivotal role of the innate immune response, particularly the dysregulated activation of pro-inflammatory cytokines and chemokines, in driving COVID-19 pathogenesis. Severe disease manifestations are often associated with a cytokine storm, resulting in widespread tissue damage and multi-organ failure.
7.1. Current therapeutic strategies and critical appraisal
Current therapeutic strategies focus on multiple targets within the viral life cycle and host immune response (Table 3). • • o o o • Major therapeutic targets and strategies in COVID-19.

7.2. Future challenges and directions
The continuous emergence of SARS-CoV-2 variants poses significant challenges, as genetic and antigenic changes may compromise vaccine efficacy and therapeutic effectiveness. This necessitates the rapid adaptation of vaccine formulations, including bivalent and potentially universal vaccines, to maintain robust protection against evolving strains. Personalized medicine approaches are increasingly recognized as vital in managing COVID-19. Identification of reliable biomarkers linked to specific inflammatory pathways could enable early risk stratification and tailored treatment regimens. Profiling patients’ immunological status, such as the balance between regulatory T cells and Th17 cells or macrophage activation states, may guide therapeutic decisions and improve outcomes. Future research should also explore the influence of comorbidities and host factors on inflammatory responses, as conditions like obesity, diabetes, and cardiovascular diseases significantly affect disease severity. Moreover, emerging evidence suggests that the microbiota may modulate host immunity and inflammation, representing an innovative frontier for intervention. Advancements in omics technologies and artificial intelligence hold great promise for unraveling the intricate virus-host interactions and identifying novel therapeutic targets. A multidisciplinary approach integrating virology, immunology, pharmacology, and clinical sciences will be indispensable to confront the evolving challenges posed by COVID-19.
8. Limitations of the study
While this review synthesizes a broad body of literature, it is subject to certain limitations. The quality of evidence varies across the cited studies, from robust RCTs to smaller observational reports and preclinical models. Our critical analysis aimed to highlight these discrepancies, but the interpretation of conflicting data remains an ongoing challenge in the field. As a narrative review, the selection and interpretation of evidence are inherently influenced by the authors’ perspectives, despite our efforts to be systematic and objective.
9. Conclusion
In conclusion, this narrative review has synthesized and critically appraised the complex immune and inflammatory pathways that define COVID-19 pathogenesis. We have underscored the dual role of the immune system, which provides essential defense but can also drive pathology through dysregulated responses like the cytokine storm. The review has highlighted key players, from innate immune cells to T cell subsets, and evaluated the evidence supporting various therapeutic targets. The continuous evolution of SARS-CoV-2 presents a persistent challenge, necessitating adaptive and multifaceted treatment strategies. Our analysis confirms that effective management of severe COVID-19 relies on a nuanced understanding of these host-virus interactions. Future efforts must focus on personalized medicine approaches and the development of broad-spectrum countermeasures to mitigate the impact of this evolving disease, as outlined in the goals of this study.
Footnotes
Acknowledgements
The authors would like to appreciate Dr Ali Hashemi because of his cooperation in removing duplications in text.
Consent to participate
This is a review article, animal and human experiments have not participated.
Author contributions
Dr. B.A and Dr A.S conceived of the presented idea. Dr E.B and Dr M.D developed the theory and Dr M.Y performed the search strategy. Dr B.A encouraged A.Y,R.N,N.B to investigate the search results and supervised the findings of this work. Dr. E.B gathered the included studies and summarized them. Dr A.S, B.A and E.B rechecked the results and critically revised the manuscript. All authors discussed the results and contributed to the final manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.