
Abstract
The severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), causes acute coronavirus disease-19 (COVID-19) that has emerged on a pandemic level. Coronaviruses are well-known to have a negative impact on the lungs and cardiovascular system. SARS-CoV-2 induces a cytokine storm that primarily targets the lungs, causing widespread clinical disorders, including COVID-19. Although, SARS-CoV-2 positive individuals often show no or mild upper respiratory tract symptoms, severe cases can progress to acute respiratory distress syndrome (ARDS). Novel CoV-2 infection in 2019 resulted in viral pneumonia as well as other complications and extrapulmonary manifestation. ARDS is also linked to a higher risk of death. Now, it is essential to develop our perception of the long term sequelae coronavirus infection for the identification of COVID-19 survivors who are at higher risk of developing the chronic lung fibrosis. This review study was planned to provide an overview of the effects of SARS-CoV-2 infection on various parts of the respiratory system such as airways, pulmonary vascular, lung parenchymal and respiratory neuromuscular system as well as the potential mechanism of the ARDS related respiratory complications including the lung fibrosis in patients with severe COVID-19.
Introduction
Coronaviruses are the positive-sense RNA viruses that infects the respiratory and gastro-intestinal system in humans and animals.1,2 The key target site of infection for the SARS-CoV-2 is the respiratory system particularly the lungs causing acute and chronic lung diseases including lung fibrosis. Patients with mild infection of COVID-19 are asymptomatic but reported positive for SARS-CoV-2, whereas those of severe infection can rapidly develop ARDS which requires mechanical ventilation and intensive care treatment. 3 Patients who survive of COVID-19 acute pneumonia are susceptible to develop harmful effects on lungs function particularly pulmonary fibrosis. 4 The occurrence of SARS-CoV-2 that can cause COVID-19, has been rapidly tuned into the pandemic, affecting many patients with underlying respiratory system disorders. 5 This review study highlights the current knowledge about the pathophysiology of COVID-19, with a particular focus on the pathogenic effects of SARS-CoV-2 on various parts of the respiratory system such as airway, vascular endothelium, alveolar epithelium, and breathing control. It also discusses the association of ARDS post SARS-CoV-2 infection with the pulmonary fibrosis and the potential mechanism underlying the lung fibrosis due to ARDS.
Literature search strategy
Relevant literature was searched from various search engines such as google scholar, Medline, Embase, directory of open access journals and Cochrane library. The key words used for the literature search were SARS, SARS-CoV-2, COVID-19 disease, COVID-19 and lungs, clinical picture and pathogenesis of COVID-19, pulmonary fibrosis, and therapeutics of COVID-19. All the research and review articles addressing the COVID-19 disease, its pathophysiology, respiratory complications in COVID-19 patients, COVID-19 therapy published during the last 5 years were included in this review study. Studies reporting COVID-19 disease co-infected with other diseases and complications other than the respiratory system were excluded from this review study. Only those articles published in English were included while articles published in any other language were excluded from this study. A total of 235 articles were retrieved through initial database searching. After removing the duplicate, 181 articles were reviewed, out of which 96 articles were assessed for eligibility while 85 articles were excluded with reasons as shown in Figure 1. Finaly, 73 articles were included in the study meeting the eligibility criteria for this review.

Prisma flow chart summary of selection process.
SARS-CoV-2 causing COVID-19
SARS-CoV-2, formerly called as nCoV 2019, is a new virus, formally reported as the major cause of a viral pneumonia outbreak on January 9, 2020. 6 Severe ARDS (SARDS) is a severe form of ARD caused by the β-CoVs family member called the CoV2, which shares 82% nucleotide similarity with human SARS-CoV and 89% nucleotide similarity with bat SARS-like CoVZXC21 and requires the angiotensin converting enzyme 2 (ACE2) receptor just like the SARS-CoV to reach the host cell.7,8 SARS-Coronavirus-2 has less genetic similarity to MERS-CoV (nucleotide identity of about 52%) 9 and causes severe respiratory disease that has a number of medical, laboratory diagnostic, epidemiologic, and radiologic manifestations resembling with the 2003 SARS-CoV infection. It appears to be transmitted predominantly from individual to individual through the droplets from respiratory system, with average 5.3 days (4.1–7.0 days 95% CI) incubation. 10
There is significant concern about the transmission of SARS-CoV-2 possibly via oral-fecal route due to the frequent diarrhea in MERS and SARS as the RNA of SARS-CoV-2 was identified in the stool specimen of SARS patients. Symptoms of gastrointestinal diseases that is, abdominal pain, vomiting, and diarrhea have been identified in 2–10% of COVID-19 patients with positive SARS-CoV-2 in fecal sample in a US patient in March 2020. 11
As of 19th March 2020, there have been 2,13,254 diagnosed COVID-19 cases with 8843 deaths worldwide. 12 The SARS-CoV-2 R0 is likely between 2 and 3, meaning that each individual infected with the virus possibly infect 2–3 others in a susceptible community. 13
The SARS-CoV-2 primarily infects adults while rarely infects younger children or children under 15 years of age.3,14–16 The global death rate was roughly 3.4%, according to the opening remarks of WHO Directors and General’s on March 3, 2020. The overall mortality rate significantly varies depending on the care, transmission intensity and geographic location. China’s overall mortality rate has been around 3.8% (6.9% in Wuhan; 0.8% elsewhere in the country). The total death rate was 1.4% in 1099 laboratory confirmed cases. During early phases of disease outbreak, China’s overall mortality rate was higher, but decreased over time which may be due to changes in patients’ quality of treatment. 17
Fever, breathlessness and cough are the three frequent symptoms of COVID-19 while less frequent symptoms include sore throat, anorexia, dyspnea, headache, and muscle pain. Symptoms may appear as soon as 3 days after exposure or to a maximum of 15 days of exposure. 12 Soon after the onset of symptoms, increased viral loads were observed, and infections in the throat were less than in the nose. 18 The SARS-CoV-2 diagnostic tests utilize the lower and upper respiratory specimens and uses real time RT-PCR as diagnostic tool in the United States. 19
Computed tomography scan of the chest evaluation is common in COVID-19 patients. Published study suggests that nearly 85% of COVID-19 patients have an abnormality on initial chest computed tomography, with 76% of patients have the involvement of bilateral lung, which manifests as ground-glass opacity in the sub-pleural and peripheral areas. 20
Pathogenesis of SARS-CoV-2-induced cytokine storm in respiratory system
Respiratory system is the main target of SARS-CoV-2 infection. The SARS-CoV-2 is an RNA β-coronavirus just like the coronaviruses that cause MERS and SARS21,22 encoding 27 proteins including 7 accessory, 4 structural, and 16 non-structural proteins. The non-structural proteins (NSP1-NSP16) are required for virus replication. 23 Its nucleocapsid is made up of two spike proteins and phosphorylated N protein, as well as genomic RNA, which is embedded in phospholipid bilayers.24,25 The two spike proteins include hemagglutinin esterase (NE) and spike glycoprotein trimmer (S), among which the envelop protein E and type III transmembrane glycoprotein M are interposed.26–28 In the lower respiratory tract, alveolar Type 2 epithelial cells exhibit the glycoprotein (S) which binds to a cell membrane receptor called ACE2. 29 The transmembrane serine protease of human type II TMPRSS2 primes S glycoprotein for this critical function, facilitating the viral entry into host cells. Cellular tropism of SARS-CoV-2 is controlled by an S1 subunit, while virus cell membrane fusion is controlled by the subunit S2. This fusion process is accompanied by the entry of genomic RNA of virus into the cytoplasm which on reaching the target is detected by the Toll-like intracellular receptor VII (TLR7) situated in endosomes. 30 As a result, RNA of the SARS-CoV-2 drives the viral proteins assembly and translation within the Golgi apparatus and ER. The recently produced vesicles holding the virus particle joins the cell membrane, thus releasing the virus. 31
The main receptor for both the SARS-CoV and SARS-CoV-2 called ACE2 allows the virus to enter target cells (such as alveolar epithelial cells), a complex immune response is triggered and characterized by the activation of Th17 lymphocytes, alveolar macrophages, CD14+ CD16+ monocytes, pathogenic Th1 cells activation. These cells produce a number of chemokines and cytokines, which contribute to the cytokine storm that maintains a “hyperinflammatory” atmosphere of neutrophils and macrophages infiltrating the lungs. In critically ill patients, the immunologically adaptive feedback driven Th1t leads to clearance of virus that appears to be lacking in COVID-19 patients suffering from ARDS (Figure 2).32,33

Thematic mechanism of SARS-CoV2 induced cytokine storm in infected lungs. 31
SARS-CoV-2 infection has been found stimulating the CD4+ T lymphocytes to develop into Th1 pathogenic cells, which secrete large number of granulocyte macrophage-colony stimulating factor (GM-CSF) and interleukin-6 (IL-6). In this type of cytokine environment, CD14+ CD16+ monocytes get activated, release IL-6, and move from the bloodstream to the lungs, where they can become macrophages of alveolar or dendritic cells (Figure 2). 32 Furthermore, critically ill patients of COVID-19 develop defective CD4+ and CD8+ T lymphocyte immunophenotypes characterized by surface markers of higher co-expression such as protein-I apoptosis and immunoglobulin T-cells and mucin containing domain-3, which predisposes to rapid exhaustion of T cells during virus infection.34,35 In fact, innate immune mechanisms may be unable to produce a targeted cytotoxic response against the virus which is normally carried out by CD8+ activated cells in severe patients. Additionally, the SARS-CoV-2-induced adaptive immune response could be characterized by a predominant Th17 profile. 36 The SARS-CoV-2 infected CD4+ T lymphocytes may be a major factor contributing to the virus induced pathogenesis and the armed T cells play important role in the prevention of pathogen infection. The T lymphocytes are the likely targets for infection caused by SARS-CoV-2. 37 It has been reported that a key subset of T cells called the regulatory T cells (Tregs) have immunoregulatory and immunosuppressive properties which play an important role in COVID-19 disease prognosis. Tregs inhibit the cells of the adaptive immune response such as T and B cells along with the inhibition of the cells of innate immune response. In COVID-19, Tregs have the ability to limit the release of excess cytokines which is the main cause of mortality and morbidity in patients infected with COVID-19. Increases in Tregs may decrease the ability of CD8+ T cells to mount an effective antiviral immune response which is critical during the initial stages of SARS-CoV-2 infection. Nonetheless, Tregs have been found to be nonfunctional or decreased in severe COVID-19 patients. 38 These T cells undergo programmed cell death leading to the dysfunctioning and depletion of T cells which eventually results lymphopenia in COVID19 patients. Furthermore, excessive inflammation triggered by the dying CD4+ T lymphocytes may lead to immunopathogenesis in COVID-19 patients. In particular, CD8+ T lymphocyte’s population is decreased significantly in COVID-19 patients. 37
In severe COVID-19, the lung pathology is described by the rapidly destroying integrity of epithelial-endothelial cells, injury to the septal capillaries, deposition of complement, accumulation of intravascular viral antigens, and restricted intravascular blood clotting. 39 The interaction of SARS-CoV-2 RNA with both the host mitochondrial protein components and the RNA results variation in size and shape of the mitochondria. 40 The above findings indicate a functional link between the host mitochondria and SARS-CoV-2, which might help the future research in understanding the pathogenic mechanisms of the viruses.
In the epithelial cells of gastrointestinal (GI) tract, higher levels of ACE2 expression is reported in GI tract than in lung. 41 ACE2 expression on the luminal epithelial cells of GI tract suggests the possible secondary target of infection by the enteric SARS-CoV-2. Symptoms such as diarrhea, vomiting, and nausea are the common GI symptoms in both the MERS-CoV and SARS-CoV-2. 42 The viral nucleocapsid protein is reported in the cytoplasm of rectal, duodenal, and gastric epithelial cells. In GI tract, SARS-CoV-2 interaction with ACE2 may lead to disrupt the claudins, occluding, barrier proteins ZO-1 and increase the production of inflammatory cytokines leading to the exacerbation of intestinal inflammation and dysbiosis. 43 ACE2 expression may exacerbate or mitigate the leaky gut in animal models. Co-morbid conditions like hypertension, obesity, and diabetes may have serious consequences on gut microbiome and SARS-CoV-2 infection and reduction in ACE2 function may excerbate the gut microbial dysbiosis. 42
Consequences of cytokine storm
In severe COVID-19 patients, the above variables suggest that cytokine storm and dysregulation have important pathological implications. The cytokine storm does, in fact, generate immunological alterations that may impair the ability of immune response to the clearance of SARS-CoV-2 disease. CD8+ and CD4+ T cells mediated protective responses of T cell against SARS-CoV-2 may fail due to excessive production of TNF-α and IL-6. T cell growth and activation suppression by these cytokines results in lymphopenia in SARS-CoV-2 severely infected patients.44,45 This notion is supported by the fact that raised TNF and IL-6 levels co-exist with low populations of CD8+ and CD4+ T cells. 34 The inhibition of functionally depleted Th1 cells has also been associated to an immunological shift towards Th2-driven responses. All of the foregoing immunological alterations are expected to develop in elderly due to the adverse effect of aging by the humoral immunity against new infections by the virus. 31
Effects of SARS-CoV-2 on airway epithelium, alveolar epithelium, and vascular endothelium
The key pathway for the entry of coronaviruses including SARS-CoV, MERS-CoV, and SARS-CoV-2 into the human is the respiratory tract epithelium, which acts like a protective barrier to pathogens, prevent infection and injury to tissues through mucus secretion and mucociliary clearing action while maintaining the efficient flow of air (Figure 3).
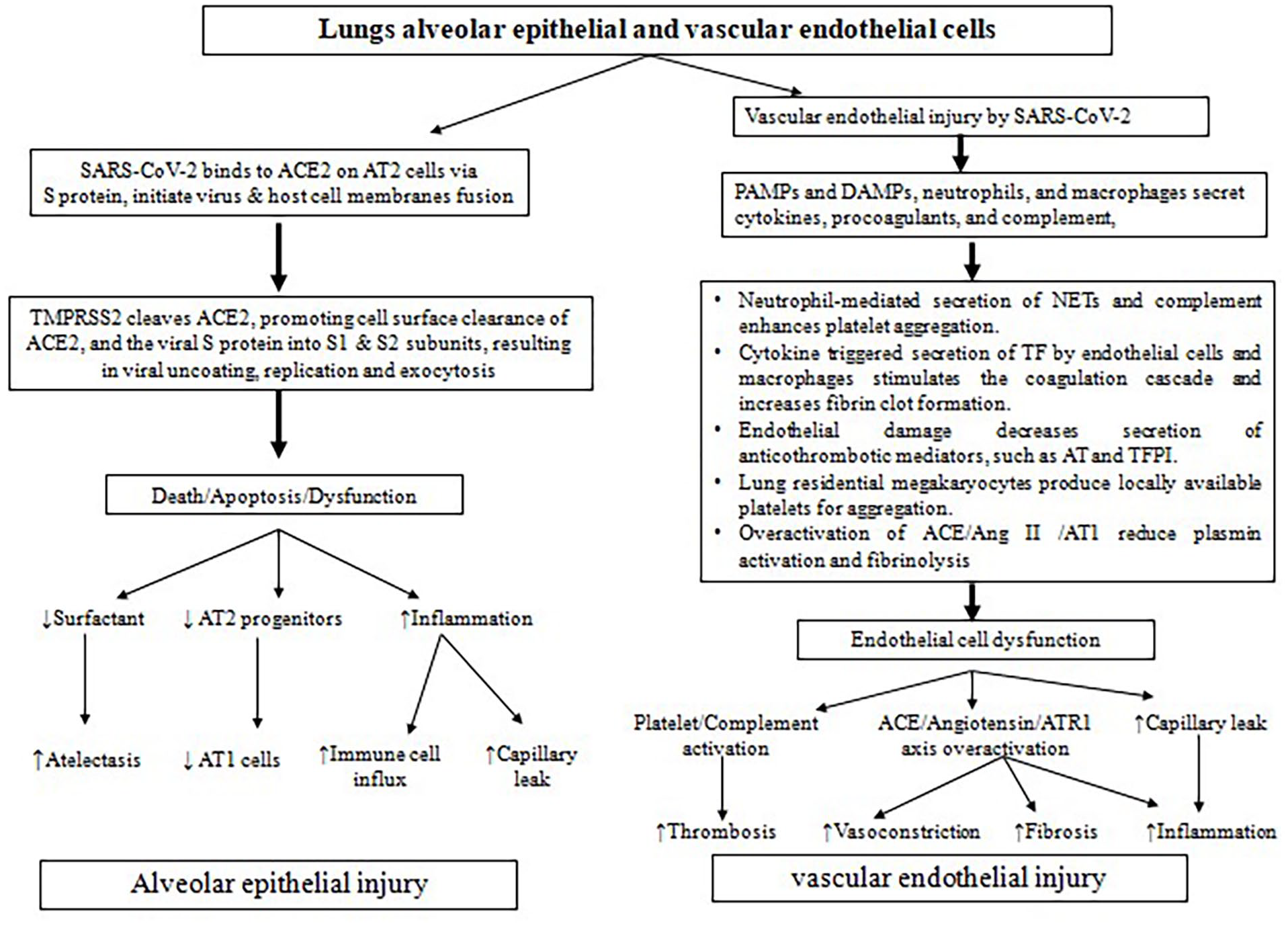
Pathophysiological mechanism alveolar epithelial and vascular endothelial injury by SARS-CoV2 infection.
Observations from the current research suggest that initially the contact of the viral particles occurs in the nasal mucosa by binding of the spike (S) protein of the virus to ACE-2 receptor, and then cleavage of the spike protein by TMPRSS2, followed by SARS-CoV-2 replication within these cells.29,46 The ACE2 protein converts angiotensin 2 to metabolites angiotensin 1-7(Ang1-7) and L-phenylalanine, many of which interfere with the renin-angiotensin-aldosterone system and exert vasodilatory properties. 47 SARS-CoV-2 travels to the airways following the entry and replication within the nasal mucosa, it triggers an inflammatory and immune response in the airways, manifesting the clinical picture of COVID-19 (Figure 3). Infected cells of the epithelium may express the mediators of inflammation such as interferons and C-X-C motif chemokine 10 (CXCL10). 48 Significant knowledge gaps exist about the SARS-CoV-2 behavior in infected ciliated cells of the epithelium of the conducting airways and whether damage to the cells results in disordered mucociliary function.
Although, the infection of SARS-CoV-2 usually begins in the upper airway epithelium, the virus diffusely infects the alveolar epithelium which results in marked impairment of gaseous exchange leading to respiratory failure.29,49 Epithelial infection has consequences other than the production of virus and loss of barrier. Coronavirus infected epithelial cells produce proinflammatory cytokines such as CXCL9, CXCL10, CXCL11, CCL5, IL-6, IL-8, and IL-29. 50 Alveolar epithelium regulates the fibrinolysis and coagulation on the surface of alveoli, mainly through urokinase and plasminogen activator inhibitor 1 (PAI1) production. 50 Pathology of SARS-CoV-2 includes hemorrhage and the deposition of fibrin in the microvasculature and alveolar space, suggesting the perturbations in clotting and fibrinolysis. It has been revealed that urokinase-related pathways predict injury to the lungs in SARS-CoV-2 infection and thus in COVID-19, alveolar epithelium and endothelium may promote coagulation disorders (Figure 3). 51
In COVID-19, the vascular disorders are a key problem and endothelium is a key target for SARS-CoV-2 infection. Vascular smooth muscle in association with pulmonary vascular endothelium effect the hypoxic vasoconstriction through the reversal of nitric oxide (NO) pathway which regulates coagulation, matches perfusion to air-space ventilation, and regulates leukocytes egress into the interstitium, a procedure mainly occurring at capillary level and is part of the barrier for solutes, water and larger molecules diffusion between the interstitial space and plasma. Considering these important functions, the loss or dysfunction of endothelium may contribute to the COVID-19 pathophysiology in lungs (Figure 3). Histopathological studies revealed that virus directly infects the endothelial cells, indicating pyroptosis, endothelial cell apoptosis, and lymphocytic inflammation of lungs and other organ’s endothelium50,52 which are associated with tissue edema, organ ischemia and a procoagulant state. 50
SARS-CoV-2 induced lung fibrosis and its potential mechanism
Pulmonary fibrosis is a well perceived abnormality of ARDS which is very common among fatal cases and is the major complication of severe COVID-19. In fatal COVID-19 cases, lung fibrosis is described by airspace obliteration and the proliferation of fibroblast which predominantly show micro-honeycombing at the autopsy. 53 The molecular mechanism responsible for profibrotic response COVID-19 is yet not completely understood. Published studies suggest that alteration in gene expression profile, direct effects of the virus, host immune response dysregulation, hypercoagulability, and local thromboembolism due to the alteration in vascular permeability, lung injury induced through mechanical ventilation may contribute to lung fibrosis induced by the SARS-CoV-2 infection54,55 (Figure 4).

Mechanism of COVID-19 associated pulmonary fibrosis. 56
ACE2 is a vital part of rennin angiotensin system (RAS) and is the main cellular receptor of SARS-CoV-2 converting Ang-II to heptapeptide Ang-(1-7), an Ang-II antagonist. Ang-II upregulates the pro-fibrotic cytokine transforming growth factor (TGF)-β1 expression causing fibrogenic effects, whereas Ang-(1-7) reduces the secretion of cytokines, decreases inflammation and protecting the lung from fibrotic remodeling and injury. 56 Previous study reported the downregulation of ACE2 expression and reduction in ACE2 activity by the SARS-CoV infection. 56 A published study reported that Ang-II levels were high in COVID-19 patients as compared to healthy individuals with positive correlation of lung injury and viral load. 57 So, the downregulation of ACE2 by the infection of SARS-CoV-2 may interfere with the RAS activities which results in lung fibrosis and inflammation.
SARS-CoV-2 spike protein contains an arginine (Arg)-glycine (Gly)-aspartic acid (Asp) integrin-binding domain mediating SARS-CoV-2 binding to integrins, including αvβ6 and αvβ3 integrins. 58 Published studies have reported that SARS-CoV nucleocapsid protein directly promotes the TGF-β signaling. Furthermore, SARS-CoV-2 binding with ACE2 improved the connective tissue growth factor (CTGF), mRNA levels of TGF-β1 and fibronectin which suggest that SARS-CoV-2 infection activate the fibrosis related genes resulting in lung fibrosis. 59
The cytokine profiling showed that severe ARDS associated patients of COVID-19 has higher levels of monocyte chemo-attractant protein-I (CCL-I), TNF, IL-17, IL-8, IL-6, IL-2, and IL-1β. 60 Inflammatory cytokines have extreme effect on the fibroblasts pro-fibrotic activities. Even though, lung fibrosis grow very slowly, the severe COVID-19 associated cytokine storm can extensively scar and thicken the small airways within a few days. 61 SARS-CoV-2 infection enhances the secretion of a key mediator of lung fibrosis called galectin-3 (Gal-3), derived by macrophages which upregulates the TGF-β receptors expression on myofibroblasts and fibroblasts through a paracrine fashion, resulting in the formation of tissue fibrosis. 62 Moreover, Gal-3 contributed in the secretion of interleukins including TNF-α, IL-6, IL-1, and IL-1β. 63 Proinflammatory cytokines like IL-1β and TNF-α causes the activation of glucuronidases that affects the intercellular junctions, degrades the endothelial cells of glycocalyx and increases the cell contractility resulting in increased permeability of vascular system and disruption in the function of endothelial barrier. 64 Altered hemostatic imbalance and vascular permeability may lead to thromboembolism and coagulopathy, which are involved in the pathogenic mechanism of pulmonary fibrosis. 56
Therapeutic implications of SARS-CoV-2 associated respiratory complications
Several medications that are currently approved for various disease treatments and work on distinct targets at molecular level may be helpful in lowering the strength of SARS-CoV-2 triggered cytokine storm.65,66 However, due to lack of published studies relevant to the efficacy profile reliable safety of used drugs in controlled trials, their application in patients infected with COVID-19 is generally empirical. A few examples of drugs include the inhibitors of JAK/STAT (Janus kinases/signal transducers and activators of transcription), azithromycin, hydroxychloroquine, corticosteroids, and the receptor antagonists of IL-6 and IL-1. 31 Corticosteroids can reduce the inflammatory response of the host in viral pneumonia thereby preventing the development of ARDS and acute lung injury. Evidence suggests that corticosteroids improved the post-COVID-19 lung fibrosis symptoms by reducing the lung inflammation and prevent the pneumonia induced by COVID-19. 67 However, serious side effects of corticosteroids usually offset its beneficial effects when used for longer period of time and at high doses, and these side effects include osteoporosis, depressive disorders, and immune depression. 68 A published study reported that dexamethasone (6 mg/day) treatment reduced the 28-day mortality of COVID-19 patients who are getting respiratory support.69,70 More clinical research is required to understand the risks and utility of corticosteroids in patients after COVID-19 infection with respiratory problems particularly lung fibrosis.
Drugs with antifibrotic potential like nintedanib and pirfenidone are efficient in promoting the reduction rate of lung function in patients with pulmonary fibrosis. Pirfenidone exerts anti-oxidative, anti-fibrotic, and anti-inflammatory properties. It has been reported that pirfenidone lowers the primary mediator of cytokine storm i-e IL-6 in serum and lungs after SARS-CoV-2 infection. It also suppresses a protein effector called furin which is involved in TGF-β1 activation, SARS-CoV-2 entry, and the signaling pathway modulation in lung fibrosis pathology following the COVID-19 infection. 71 Azithromycin can maintain the integrity of epithelial cell, downregulate the production of cytokine, and prevent the entry and binding of SARS-CoV-2 into the interstitial cells of lungs, suggesting its inhibitory effects against lung fibrosis induced by COVID-19 through its anti-viral and immunomodulatory properties. 72 Another potential treatment option for pulmonary fibrosis is the mesenchymal stem cells (MSCs) therapy and its antifibrotic effects are linked to their angiogenic, anti-inflammatory and immunosuppressive properties. MSCs have the ability to regenerate and reinstate the structure and functions of healthy lung tissue. The MSCs secretome have micro/nanostructured extracellular vesicles (EVs) and soluble proteins (growth factors, chemokine and cytokine), which through antifibrotic and anti-inflammatory activities may mediate the fibrotic lung regeneration.73,74
To control COVID-19, extensive research is in process of developing the antivirals. A broad spectrum antiviral called remdesivir, a nucleotide analog when used with chloroquine has the inhibitory effect against SARS-CoV-2 infection. Remdesivir is under evaluation for both mild to moderate (NCT04252664) and severe (NCT04257656) COVID-19 infections that was originally developed for treating Ebola virus infection, but proved ineffective. 75
The effect of azithromycin and hydroxyl chloroquine has been tested on respiratory viral loads in COVID-19 patients through a limited, small, and single-arm French study. Sixteen patients included in the study left untreated and served as controls while 20 patients were treated with hydroxyl chloroquine. Azithromycin was also given to six patients already treated with hydroxyl chloroquine for superimposed infection. On day 6, significant reduction in viral load was found in hydroxyl chloroquine treated patients compared to control individuals. 76 More effective and faster elimination of virus was observed in six patients who were treated with both the azithromycin and hydroxyl chloroquine. 77
A 15 day regimen of ritonavir lopinavir was compared to usual treatment in 199 hospitalized COVID-19 patients with decreased indices of oxygen saturation in a randomized, monitored, open-label trial. Lopivir-ritonavir did not improve clinical outcomes, decrease 28 day mortality, or diminish the detectability of viral RNA in throat as compared to standard therapy. 78 Table 1 shows a variety of drugs that are being evaluated or are being considered for testing the patient of coronavirus-19. 79
An overview of selected clinical trials registered for COVID-19 treatment. 79 .

CTLs: cytotoxic T cells; COVID-19: Coronavirus disease 2019; PD-1: programmed cell death-1.
Limitations
The present review study on the fundamentals of COVID-19 and other coronaviruses focusing on the pathogenesis and the implication of COVID-19 disease on the respiratory system of the infected patients have certain limitations. Although, data presented in this review covers most of the aspects of COVID-19 disease particularly focusing on the vulnerability of respiratory system with the virus leading to lung fibrosis but the collected data used in this review is taken from a diverse literature of published studies that make it very difficult to compile a comprehensive narrative review. Comorbidity of COVID-19 disease with other life-threatening diseases affecting the vital organs of the body make it highly complex. It is critical to design the future research to identify the high-risk patients of COVID-19 who will be more prone to other life-threatening disorders particularly the respiratory disorders.
Conclusions
The continued improvement in our understanding of underlying pathogenic pathways is being related to the recent dramatic epidemic of SARS-CoV-2 infection, which is providing some light on the immunophenotypic features that make infected individuals more vulnerable to cytokine storm-induced lung damage. Indeed, a weakened antiviral immune response combined with an abnormal hyperinflammatory response can make the most serious types of coronavirus-19-related illness more likely, particularly in older ages with comorbidity. As the transmission path of the disease and the new data becomes available, it will be wise to establish the risk factors for the development of respiratory problems in COVID-19 patients. It’s also possible to look into the therapeutic efficacy of JAK/STAT inhibitors and IL-6 receptor antagonists. There are considerable evidences that critical patients of COVID-19 develop fibrotic sequelae related to the ARDS and the functional impairment of the lungs indicate restrictive respiratory complications. Currently, there is no treatment guidelines and long-term studies to mitigate COVID-19 associated lung fibrosis. It is critical to design studies to identify high risk patients developing the long-term pulmonary fibrosis and its impact on lung function.
Footnotes
Abbreviations
SARS-CoV-2 severe acute respiratory syndrome coronavirus-2
COVID-19 coronavirus disease-19
ARDS acute respiratory distress syndrome
RNA ribonucleic acid
nCoV novel coronavirus
MERS middle east respiratory syndrome
WHO world health organization
RT-PCR reverse transcriptase-polymerase chain reaction
TMPRSS2 transmembrane serine protease of human type II
TLR7 toll-like intracellular receptor VII
ACE angiotensin converting enzyme
NSP nonstructural protein
GM-CSF granulocyte macrophage-colony stimulating factor
IL-6 interleukin-6
Tregs regulatory T cells
GI gastrointestinal tract
TNF-α tumor necrosis factor- α
TNF tumor necrosis factor
PAI1 plasminogen activator inhibitor 1
NO nitric oxide
RAS rennin angiotensin system
TGF transforming growth factor
CTGF connective tissue growth factor
MSCs mesenchymal stem cells
EVs extracellular vesicles
Author’s contribution
GR, AMK, MR, SA contributed in study design, supervision, literature search, manuscript draft writing, and critical review. WAK, MA, AR, SI contributed in literature search, manuscript drafting, and construction of figures and table.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical statement
Our study did not require an ethical board approval because it is not applicable.
Informed consent
Not applicable