
Abstract
The objective of this study was to investigate the effects of Cervus nippon var. mantchuricus water extract treated with digestive enzymes (CE) on the promotion of M1 macrophage polarization in murine macrophages. Macrophages polarize either to one phenotype after stimulation with LPS or IFN-γ or to an alternatively activated phenotype that is induced by IL-4 or IL-13. Cell viability of RAW264.7 cells was determined by WST-1 assay. NO production was measured by Griess assay. IL-6, IL-12, TNF-α, and iNOS mRNA levels were measured by RT-PCR. IL-6, IL-12, and IL-10 cytokine levels were determined by ELISA. TLR4/MAPK/NF-κB signaling in RAW264.7 cells was evaluated by western blotting. The level of NF-κB was determined by immunoblotting. CE induced the differentiation of M1 macrophages. CE promoted M1 macrophages to elevate NO production and cytokine levels. CE-stimulated M1 macrophages had enhanced IL-6, IL-12, and TNF-α. CE promoted M1 macrophages to activate TLR4/MAPK/NF-κB phosphorylation. M2 markers were downregulated, while M1 markers were upregulated in murine macrophages by CE. Consequently, CE has immunomodulatory activity and can be used to promote M1 macrophage polarization through the TLR4/MAPK/NF-κB signaling pathways.
Introduction
Macrophages respond to environmental cues in a polarized manner. 1 Macrophages polarize either to one phenotype after stimulation with lipopolysaccharide (LPS) or interferon (IFN)-γ or to an alternatively activated phenotype that is induced by interleukin (IL)-4 or IL-13. 2 Classically activated macrophages (M1) are characterized by their expression of cytokines such as IL-12 and IL-6. Alternatively, activated macrophages (M2) typically express arginase-1, which is also termed chitinase-like 3. 3 Macrophages convert phenotypes according to the tissue environment, and this phenotypic plasticity is beneficial to both the host and pathogens.4,5 In addition, inappropriate host reactions can cause chronic wounds and pathogen invasion, which can lead to tissue damage.
Cell-surface protein Toll-like receptor 4 (TLR4) reacts to lipopolysaccharide (LPS) as a member of the TLR family involved in the innate immune system and interacts with MyD88.6–8 LPS activates macrophages and plays a major role in immune and inflammatory processes.9,10 These cells are induced by TLR ligands and secrete numerous cytokines such as tumor necrosis factor (TNF)-α, IFN-γ, IL-1β, IL-6, and IL-12, and nitric oxide (NO), inducible NO synthase (iNOS) and prostaglandin E2 (PGE2).11,12 In particular, the mitogen-activated protein kinase (MAPK) and nuclear factor (NF) -κB signaling pathways are key intracellular transduction cascades involved in differentiation, cell activation, and cytokine secretion of macrophages.13,14
Enzyme hydrolysis is used as a diet for people or patients with metabolic problems such as glycoproteins, proteins, and lipids, which are polymers. Especially, alcalase is a crude protein produced for commercial, which is a non-special serine-type endogenous protein, and it is widely used in the food industry as a hydrolysed protein that is more nutritious or functional than intact proteins.15,16
The bones of various animals, including deer and chickens, have been used as traditional medicines, are still used today, and have been studied. In particular, deer bone extract, also known as water extract from Cervus nippon var. mantchuricus (CM), is the most commonly used Asian medicine. Currently, CM, supplied by Nong Shim Corporation and Food Company, is sold in the market as coffee and healthy drinks. Previous studies have indicated that oral CM administration is useful for the treatment of various diseases (inflammatory diseases, atopic dermatitis, memory loss, bone resorption, and neutropenia).17–22 However, there is no study on the effect of Cervus nippon var. mantchuricus water extract treated with digestive enzymes (CE) on M1 macrophage polarization.
Our data clearly show that CE can be used to promote M1 macrophage polarization through the TLR4/MAPK/NF-κB signaling pathways, and assessed the immunomodulatory activity of CE in vitro in macrophages.
Methods
Preparation of CE
CE was prepared by Nong Shim Corporation and Food Company (Seoul, Korea). Dried CE powder was stored in aliquots at −80°C until further analysis (Table 1). Alcalase, the digestive enzyme used in this study, was purchased from Novozymes Co., Ltd. (Denmark) and its specification, optimal pH and temperature, and protease activity are shown in Table 2. The protease activity of enzymes was measured by substrate of casein according to the methods of Senphan and Benjakul, 23 and the amount of enzymes needed to produce tyrosine of 1 μmol per unit time (min) was determined in units of 1 unit under optimal conditions. Detailed hydrolysis-related condition and additional information on CE production processes may be requested from the company (http://nongshim.co.kr).
The composition of CE.

The specification, optimal pH, and temperature for activity of digestive enzymes.

One unit of protease activity means as the amount of enzyme that produces 1 μmol of tyrosine from 0.1% casein per min under the optimal conditions.
Cell culture and morphology
RAW 264.7 cells, a murine monocyte/macrophage cell line, were purchased from the Korea Cell Line Bank (KCLB no. 40071, Seoul, Korea). The cell line was grown in Dulbecco’s modified Eagle’s medium (DMEM, Welgene, Daegu, Korea) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Welgene, Daegu, Korea) and 1% antibiotics (Ab, Welgene, Daegu, Korea) at 37°C in a 5% CO2 humidified incubator. The cells were divided into five groups: (1) M0: RAW 264.7 cells cultured with regular medium; (2) M1: RAW 264.7 cells stimulated with 1 μg/ml LPS; (3) CE 10: RAW 264.7 cells cultured with 10 mg/ml CE; (4) CE 50: RAW 264.7 cells cultured with 50 mg/ml CE; and (5) CE 200: RAW 264.7 cells cultured with 200 mg/ml CE. Each group was cultured for 24 h prior to the following experiments. RAW 264.7 cells (5 × 105 cells/ml) were cultured to induce M1 polarization in the presence of LPS (1 μg/ml; Sigma, EMD Millipore, Billerica, MA, USA) and CE (10, 50, 200 mg/ml) for 24 h in 6-well plates. Morphological changes were analyzed by taking images using a camera connected to a light microscope (Olympus, Tokyo, Japan). We followed the methods of Hong et al. 24
Detection of NO
Nitric oxide (NO) production was determined in RAW 264.7 culture supernatant using a Griess reagent kit (Promega, Madison, WI, USA). In brief, 150 µl of culture supernatant was transferred to a 96-well plate and then mixed with 150 µl of Griess reagent solution. Mixtures were then incubated for 30 min at room temperature. Optical density was measured at 570 nm using a microplate reader. (Versa Max; Molecular Devices LLC, Sunnyvale, CA, USA).
Cell viability assay
Cell viability was measured using a WST-1 assay (Dogen, Seoul, Korea). RAW264.7 cells (1 × 104 cells/well) were plated in 96-well culture plates and incubated for 24 h. Cells were then treated with different concentrations of CE and incubated for another 24 h. Then, 10 µl WST-1 solution was added to 100 µl cell culture medium, and the plates were incubated for 40–60 min. The optical density was determined at 450 nm using an enzyme-linked immunosorbent assay (ELISA) reader (Versa Max; Molecular Devices LLC, Sunnyvale, CA, USA).
Western blot analysis
Cells were harvested, and cell pellets were incubated in one volume of lysis buffer containing 20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 1 mM Na3VO4, 1 mM DTT, 1 mM NaF, 1 mM PMSF, and PI cocktail on ice for 30 min and centrifuged at 13,000 rpm for 20 min at 4°C. Aliquots containing 20 µl nitrocellulose membranes (Protran nitrocellulose membrane, Whatman, UK). Membranes were blocked with 5% nonfat milk, probed with specific primary antibodies, and transferred to nitrocellulose membranes (Protran nitrocellulose membrane, Whatman, UK). After washing in 1X PBST for 15 min, the membranes were incubated with diluted enzyme-linked secondary antibodies, and after washing in 1X PBST for 1 h, the protein bands were detected using the EZ-western chemiluminescent detection kit and visualized by exposing the membranes to X-ray films. In a parallel experiment, cytoplasmic and nuclear proteins were extracted using NE-PER® nuclear and cytoplasmic extraction reagents (Pierce Biotechnology, Rockford, IL, USA) according to the manufacturer’s instructions. Each protein was blotted with the appropriate antibodies. Anti-TLR4, anti-NF-κB, anti-p-ERK1/2, anti-p-P38, anti-p-JNK, and anti-Lamin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-iNOS, anti-COX-2, anti-p-NF-κB, and anti-GAPDH antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA).
Immunofluorescence (IF) assay
RAW 264.7 cells were fixed with 4% paraformaldehyde in 0.1 M PBS for 15 min. After rinsing with PBS, the coverslips with adherent cells were used for immunofluorescence staining. In each group, RAW 264.7 cells were incubated with anti-mouse TLR4 (1:100; Santa Cruz, Biotechnology, Inc., Dallas, Texas, USA) overnight at room temperature. Subsequently, the cells were incubated in anti-Alexa Fluor-488 secondary antibody (Invitrogen, Eugene, Oregon, USA) for 1 h at room temperature. After washing, the coverslips were mounted using a fluorescent mounting medium with 4,6-diamidino-2-phenylindole (DAPI; Sigma, EMD Millipore, Billerica, MA, USA). Images were obtained with an Olympus FV10i self-contained confocal laser system (Fluoview1000, Olympus, Tokyo, Japan). The object was 40×, and scale bars on the image indicate 20 μm.
Enzyme-linked immunosorbent assay (ELISA)
The expression of cytokines (IL-6, IL-12, and IL-10) by RAW264.7 cells was measured by sandwich ELISA using the BD Pharmingen mouse or human ELISA set (BD Biosciences, San Diego, CA, USA). In detail, the plates were coated with capture antibody in ELISA coating buffer (Sigma, EMD Millipore) and incubated overnight at 4°C. The plates were washed with PBS with 0.05% Tween 20 and subsequently blocking buffer (10% FBS in PBS) for 1 h at 20°C. Serial dilutions of standard antigen or sample in dilution buffer (10% FBS in PBS) were added to the plates, and the plates were incubated for 2 h at 20°C. After washing, biotin-conjugated anti-mouse cytokines (IL-6, IL-12, and IL-10) (1:500 dilution) and streptavidin-HRP conjugate (1:250 dilution) were added to the plates, and plates were incubated for 1 h at 20°C. Finally, tetramethylbenzidine substrate solution was added to the plates, and after 20 min of incubation in the dark, a 1 M H3PO4 solution was added to stop the reaction. OD was measured at 450 nm on an automated ELISA reader (VersaMax; Molecular Devices LLC).
Reverse transcription-polymerase chain reaction (RT-PCR)
The cells were harvested by centrifugation (500 × g, 20 min, 4°C), and the pellet was washed with ice-cold PBS. RNA was isolated using the Easy-Blue RNA extraction kit (iNtRON Biotech, Sungnam, Korea) according to the manufacturer’s protocol. The isolated RNA content was measured using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE, USA). Next, 2 µg of total cellular RNA from each sample was reverse transcribed using a cDNA synthesis kit (Takara Bio, Inc., Otsu, Japan). PCR was conducted in a 20 µl reaction mixture consisting of DNA template, 10 pM of each gene-specific primer, 10× Taq buffer, 2.5 mM dNTP mixture, and 1 unit of Taq DNA polymerase (Takara Bio, Inc., Otsu, Japan). PCR was performed according to the instructions of the Takara Taq kit as follows: pre-DNA denaturation at 95°C for 3 min; 30 cycles of DNA denaturation at 95°C for 45 s; annealing for 40 s at 56°C; and elongation at 72°C for 50 s. The PCR was performed using a SimpliAmp thermal cycler (Applied Biosystems; Thermo Fisher Scientific, Inc.). The PCR products were subsequently separated using agarose gel electrophoresis [5% (w/v)] and were stained with ethidium bromide (room temperature for 5 min). All experiments were performed in triplicate. The relative optical density ratio was calculated using ImageJ software (version 1.42q; National Institutes of Health, Bethesda, MD, USA) with GAPDH as an internal control. The primer sequences used for mouse iNOS, IL-6, TNF-α, IL-10, and GAPDH are shown in Table 3.
The sequence of PCR primer.

Data analysis
All experimental results are represented as the means ± SEM of at least three separate tests. Statistical significance was determined using a one-way ANOVA and Tukey-Kramer multiple comparisons posttests to analyze differences between groups. Statistical significance at P < 0.05, <0.01, and <0.001 were given respective symbols in the figures. Statistical analyses were performed using PRISM software (version 5.0; GraphPad Software Inc., La Jolla, CA, USA).
Results
CE stimulated M1 macrophage cell polarization
We relied on established protocols to polarize RAW264.7 macrophages into M1 macrophages by exposure to LPS. 25 To determine the effect of CE on cell viability, RAW264.7 cells were treated with LPS or various concentrations of CE for 24 h. CE did not induce any cytotoxicity in RAW264.7 cells over a 24 h period. Moreover, both CE and LPS increased WST activities in RAW264.7 cells, indicating that CE induced dehydrogenase activities (Figure 1(a)). Thus, the macrophages were treated with CE at concentrations of 10, 50, and 200 μg/ml. Next, we investigated the morphology of the macrophages with LPS or CE treatment (Figure 1(b)). LPS has been known to stimulate the differentiation of monocytic macrophages to M1 macrophages. 12 As a result, LPS stimulated macrophage differentiation in our experiment. Similarly, CE treatment also resulted in macrophage cell differentiation in a dose-dependent manner. Consistently, these results demonstrate that CE induces M1 macrophage phenotype polarization.
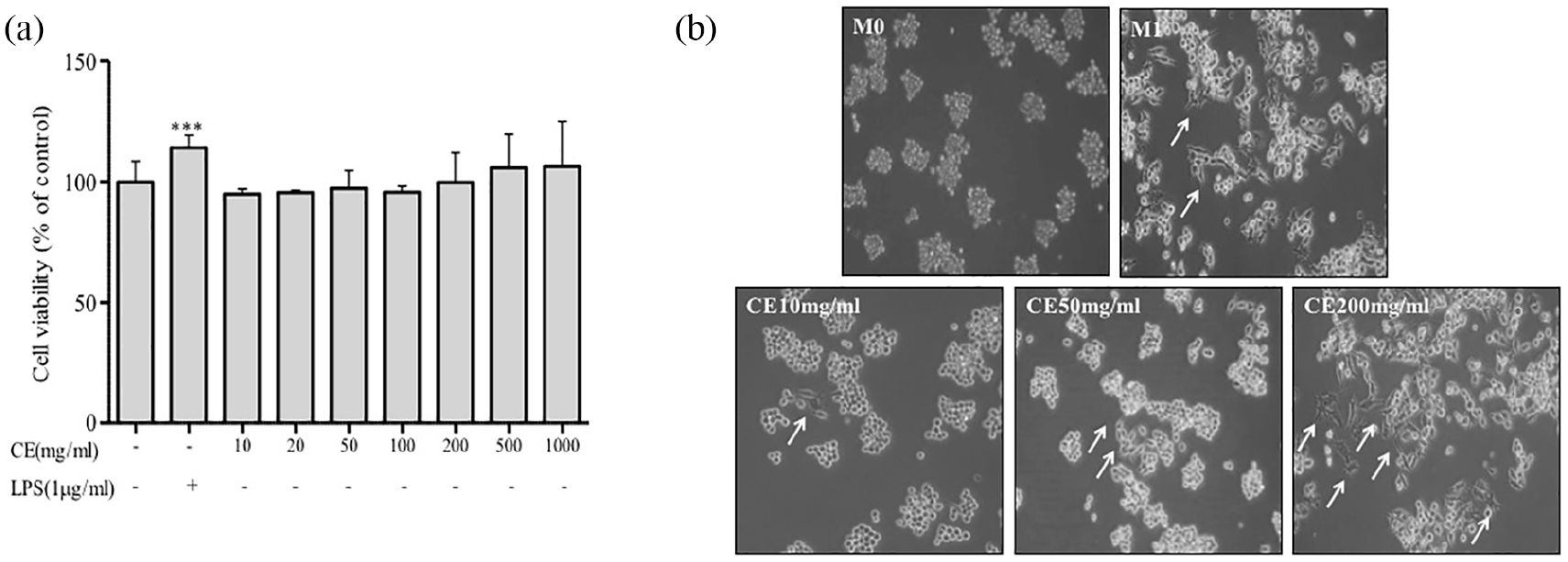
CE activates macrophage cells. RAW264.7 cells (1 × 104 cells/ml) were treated with various concentrations of CE or LPS (1 μg/ml) and then incubated for 24 h: (a) cell viability was measured using a WST assay and (b) cells were seeded onto 60 mm culture dishes at a density of 3 × 104 cells/dish. The next day, cells were treated with CE for 24 h. RAW 264.7 cell surfaces were observed by taking photographs using a camera attached to a microscope.
CE induced M1 macrophages to elevate NO production
To test the ability of RAW264.7 cells to respond to a known inducer of the M1 phenotype, we measured the gene expression of the M1 marker iNOS in response to LPS. 2 As a result of RT-PCR analysis, iNOS mRNA levels increased like M1 due to CE treatment in RAW264.7 cells (Figure 2(a)). Similarly, CE-mediated stimulation of M1 macrophages significantly promoted NO production (Figure 2(b)). Moreover, CE treatment increased the expression of iNOS (Figure 2(c)). Our findings indicate that CE enhances the activation of mRNA (iNOS) levels and NO production, consistent with M1 macrophages, in RAW264.7 cells.
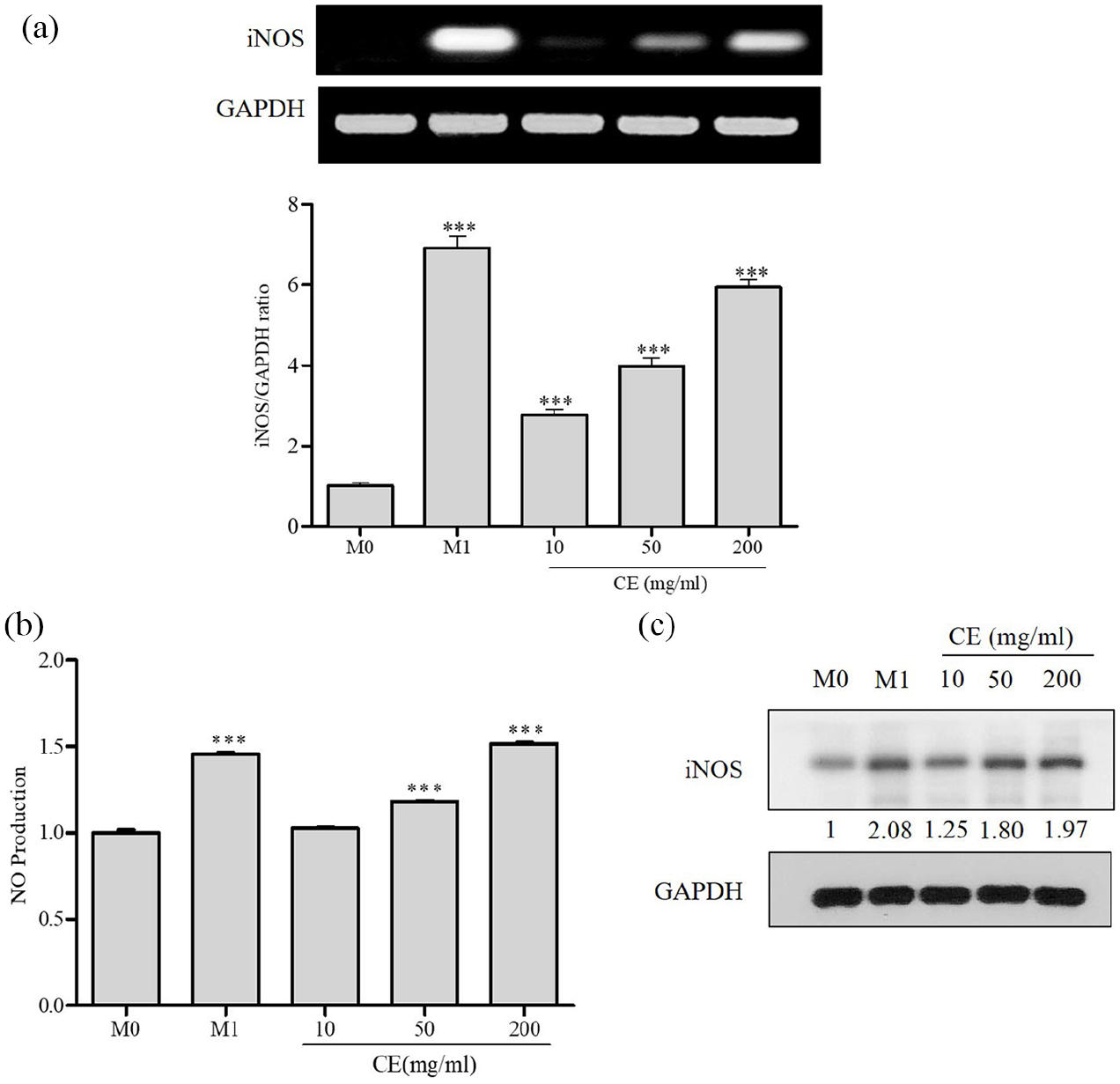
CE-induced M1 macrophage polarization regulates iNOS and NO production. RAW 264.7 cells were stimulated with CE (10, 50, or 200 mg/ml) or LPS for 24 h: (a) iNOS mRNA levels were measured by RT-PCR analysis. The bar graphs represent the quantitation of the RT-PCR data, (b) the level of NO production in cell culture supernatants was measured by sandwich ELISA and (c) isolated iNOS from cell lysates was determined by immunoblotting. GAPDH was used as an internal control.
CE promoted M1 macrophage polarization to enhance IL-6, IL-12, and TNF-α
Next, we examined whether CE has a role in the production of cytokines by M1 macrophages stimulated with LPS. It is well known that LPS interacts with TLR4 and stimulates murine macrophages. 7 M1 macrophages are generally characterized as IL-6high, IL-12high, TNF-αhigh, and IL-10low. 26 As observed, LPS-mediated stimulation of M1 macrophages considerably promoted the expression of IL-6, IL-12, and TNF-α. As a result of the RT-PCR analysis, the CE treatment of RAW264.7 cells increased IL-6 and TNF-α mRNA levels, as in M1 (Figure 3(a)). The upregulation mRNA levels of immunostimulating M1 macrophage-associated cytokines (IL-6 and IL-12) was discovered in the RAW264.7 cells treated with CE compared with the levels in the M0 group (Figure 3(b)). Under these conditions, we measured lower levels of IL-10 expression in the RAW264.7 cells treated with CE compared with those of the M0 group (Figure 3(c)). Furthermore, we also found that CE enhanced the activation of mRNA (IL-6 and TNF-α) levels and cytokine levels (IL-6 and IL-12), similar to M1 macrophages, in RAW264.7 cells.

CE-induced M1 macrophage polarization regulates mRNA expression (IL-6, TNF-α, and IL-10) and cytokine levels (IL-6 and IL-12). RAW 264.7 cells were stimulated with CE (10, 50, or 200 mg/ml) or LPS for 24 h: (a) the IL-6 and TNF-α mRNA levels were measured by RT-PCR (upper panel). The bar graphs represent the quantitation of the RT-PCR data (lower panel), (b) the levels of IL-6, IL-12, and IL-10 in cell culture supernatants were measured by sandwich ELISA, (c) the IL-10 mRNA levels were measured by RT-PCR. The bar graphs represent the quantitation of the RT-PCR data and (D) the level of IL-10 in cell culture supernatants was measured by sandwich ELISA.
CE promoted M1 macrophage polarization activation via TLR4/MAPKs/NF-κB pathways
To establish the mechanism by which CE initiates cytokines (IL-6, IL-12, and TNF-α) and upregulates NO production, we examined the TLR4, MAPKs, and NF-κB signaling pathways. LPS treatment resulted in activation of the MAPKs and NF-κB pathways via TLR4. 27 TAK-242 (TLR4i; TLR4 inhibitor) is an exogenous synthetic selective inhibitor of TLR4, thereby inhibiting TLR4-mediated downstream signaling events. 28 When RAW264.7 cells were treated with CE for 24 h, TLR4 and ERK phosphorylation were marginally increased with 50 and 200 µg/ml CE treatment, similar to M1 stimulation of TLR4. Additionally, we showed that CE activates phosphorylation of JNK and p38. JNK and p38 phosphorylation were slightly increased after treatment with 50 and 200 µg/ml CE (Figure 4(a)). CE caused the phosphorylation of ERK, JNK, and p38 in a dose-dependent manner. Thus, CE treatment resulted in nuclear and cytosolic accumulation of the phosphorylated form of NF-κB (Figure 5(a)). As a result, we utilized TLR4i to examine the role of TLR4 in CE-induced MAPKs (p38, JNK, and ERK)/NF-κB phosphorylation. As shown in Figures 4(b) and 5(b), the expression of TLR4/MAPKs/NF-κB phosphorylation was suppressed by TLR4i. This result was further confirmed by immunocytochemistry, as shown in Figure 4(c). Our immunofluorescence data indicate that TLR4i combined with 200 mg/ml CE or M1 stimulates TLR4 in the cytosol in RAW 264.7 cells (Figure 4(c)). The immunofluorescence results were consistent with western blot analysis. The translocation of TLR4 from the cell membrane to the cytosol was increased in the cells treated with 200 mg/ml CE or LPS-stimulated M1 compared to that of the untreated control (M0). In addition, pretreatment with TLR4i almost completely abolished the 200 mg/ml CE- or M1-induced translocation of TLR4. These data collectively demonstrate that CE activates the TLR4 pathway in RAW264.7 cells. However, it remains unknown whether the phosphorylation of MAPKs by CE is mediated by TLR4 and related to M1 macrophage cytokines. The immunostimulating activities of CE observed in macrophages were further confirmed by examining the effects of CE on macrophages. M1 macrophages are generally characterized as IL-6high and IL-10low. Our data showed that TLR4i inhibited the CE- or M1-induced expression of IL-6 (Figure 5(c)). Moreover, we measured lower levels of IL-10 expression in cells pretreated with TLR4i, followed by treatment with CE or M1 compared with that of the M0 group (Figure 5(d)). Taken together, CE upregulates TLR4 surface expression, followed by stimulation of MAPKs, resulting in NF-κB translocation into the nucleus. Our findings indicate that CE induces the activation of TLR4/MAPK/NF-κB pathways, similar to the M1 phenotype.

CE-induced TLR4/MAPK signaling activation in macrophages: (a) cells were stimulated with CE (10, 50, or 200 mg/ml) or LPS (1 μg/ml) for 24 h. Phosphorylated ERK, p38, JNK from cell lysates were determined by immunoblotting, (b) cells were prestimulated for 2 h with TLR4i (1 μM) and then stimulated with CE (200 mg/ml) or LPS (1 μg/ml) for 24 h. Phosphorylated ERK, p38, JNK from cell lysates were determined by immunoblotting. GAPDH was used as an internal control and (c) cells were preincubated for 2 h with TLR4i (1 μM), then incubated with CE (200 mg/ml) or LPS (1 μg/ml) for 24 h, and observed by laser scanning confocal microscopy (40×). Representative images and results from three independent experiments are shown.

CE-induced NF-κB signaling activation in macrophages: (a) cells were stimulated with CE (10, 50, or 200 mg/ml) or LPS (1 μg/ml). Cytoplasmic extracts (left panel) and nuclear extracts (right panel) were isolated from cells stimulated with CE or LPS. The level of NF-κB was determined by immunoblotting, (b) cells were prestimulated for 2 h with TLR4i (1 μM) and then stimulated with CE (200 mg/ml) or LPS (1 μg/ml) for 24 h. The level of NF-κB was determined by immunoblotting. Tubulin and lamin were used as loading controls for the cytoplasmic and nuclear fractions, respectively, (c) the level of IL-6 in cell culture supernatants was measured by sandwich ELISA and (d) the level of IL-10 in cell culture supernatants was measured by sandwich ELISA.
Discussion
The objective of this study was to characterize the M1 polarization immunomodulatory activity of CE via the TLR4 signaling pathway by performing experiments in macrophages.
Macrophages are heterogeneous in phenotype and exhibit plasticity in polarizing to adapt to different tissue environments. Two types of macrophages have been identified in infection and tissue repair, namely, classically activated macrophages (M1) and alternatively activated macrophages (M2). M1 macrophages are usually associated with increased proinflammatory cytokine production, which initiates adaptive immune T cell responses and induces tissue inflammation by producing cytokines.29–31
LPS can induce inflammation and promote M1 macrophage polarization. Moreover, it is now apparent that the Toll-like receptor (TLR) family plays an important role in the direct activation of host defense mechanisms. The activation of TLRs stimulates the innate immune response accompanied by NO production, 32 and increases the adaptive immune response by inducing the generation of various cytokines, such as IL-1β, IL-6, TNF-α, IFN-γ, and IL-12, which increase both cell-mediated and immune responses. 33
The purpose of this study was to focus on the immunomodulatory activity of CE. For that purpose, cell differentiation was discovered and cytokine, mRNA, and NO production levels associated with immunomodulatory activity were measured. Our results show that CE increases differentiation due to M1 macrophage polarization and increases production of effector molecules such as immunostimulation cytokine (IL-6, TNF-α, and IL-12) and NO. In this study, CE raised the mRNA levels of iNOS, TNF-α, and IL-6 through M1 macrophage polarization, and as with LPS, it appears to have a similar effect on cell differentiation of macrophages and cytokine production.
Previous studies have shown that stimulation of TLRs leads to the activation of MAPK pathways, resulting in the increase of antibody and cytokine production in M1 macrophages, and that the MAPK pathways play an important role in innate immune response signaling.34,35 The major families of the MAPK pathways that mediate innate immune response signaling include ERK1/2, p38, c-Jun N-terminal kinase (JNK), and the activation of the MAPK pathway subsequently induces gene expression by activating typical transcription factors such as NF-κB.36,37 NF-κB activity in macrophages can be caused by various factors stimulating LPS, and transcription factors containing p65 activated in nucleus are closely related to regulating the expression of genes associated with immune responses such as TNF-α and inducible nitric oxide synthase (iNOS).38–40 We found that CE activated TLR4/MAPKs (ERK1/2, P38, and JNK) phosphorylation via M1 macrophage polarization. Therefore, the downstream regulators of the NF-κB signaling pathway might be the targets for CE. Thus, CE induced differentiation into M1 macrophages through modulation of MAPK and NF-κB transcription factors, and showed excellent response to the expression of immune-enhancing cytokines. Our results demonstrate that CE can selectively and efficiently activate M1 macrophages. Notably, we detected that CE enhanced M1 macrophage activation markers. As a result, CE upregulates TLR4 surface expression, then stimulates MAPK to translocate NF-κB to the nucleus, and these results support the conclusion that CE promotes M1 macrophage polarization through the TLR4 signaling pathway.
Conclusion
Taken together, CE promotes M1 macrophage polarization. CE induced the activity of M1 macrophage polarization and the expression of immunomodulatory cytokine mRNA and increased the levels of NO. Most importantly, CE contributed to its immune-enhancing effect by stimulating M1 macrophages through the upregulation of TLR4/MAPKs/NF-κB signaling pathways. These results support the feasibility of CE as a good candidate compound for the activation of TLR4 signaling and the promotion of M1 macrophage polarization.
Footnotes
Author contributions
SHH carried out the experiment and drafted the manuscript. SHH, JMK, YSL, and HIK revised the research and manuscript and assisted in the research work. SHH guided the research and revised and submitted the manuscript. S-GK supervised the research. All the authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the Korean Medicine Research & Development Project of the Ministry of Health and Welfare (grant numbers HI12C1889, HI13C0530, HI18C2382, and HI11C2110) and Nong Shim Corporation (grant number 20121086). The funding sponsors had no role in the study design, performance, data collection and analysis, decision to publish, or preparation/writing of the manuscript.