
Abstract
The purpose of this study was to identify the mechanism of lipopolysaccharide (LPS)-induced expression of tumor necrosis factor (TNF)-α in BEAS-2B. Toll-like receptor (TLR)4-specific siRNA was found to completely abolish the LPS-induced expression of MyD88 and TNF-α. There was enhanced binding of MyD88 with IRAK1 following LPS treatment, and MyD88- or IRAK1-specific siRNAs decreased the expression of TNF-α. In addition, IRAK1 siRNA downregulated the phosphorylation of PKCα, demonstrating that PKCα is a downstream effector of IRAK1. Inhibition of PKCα completely blocked the activation of AKT, whereas inhibition of AKT with a PI3K inhibitor prevented the LPS-induced expression of TNF-α. We found that AKT activated JNK, which then stimulated phosphorylation of Iκ-Bα, resulting in NF-κB activation. As expected, inhibition of NF-κB completely inhibited the expression of TNF-α. Taken together, our results suggest that LPS induces TNF-α expression by activating NF-κB via the PKCα/PI3K/AKT/JNK pathway, which is in turn dependent on MyD88/IRAK1.
Introduction
Inflammation is part of a complex response of vascular tissues to chemical or mechanical injury, and infection.1,2 Bacterial lipopolysaccharides (LPS), a major constituent of the outer membrane of many Gram-negative bacteria, has been identified as a crucial agent of inflammation. 1 Exposure of mammalian cells to LPS stimulates the release of pro-inflammatory cytokines, which in turn activate a second set of inflammatory cascades involving cytokines, lipid mediators, and adhesion molecules. 3 Stimulation with LPS leads to increased expression of TLR4, and triggers the activation of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and interleukin-1β (IL-1β) in macrophages. 4
Toll-like receptors (TLRs) detect microorganisms and protect the host against infection by inducing the release of inflammatory cytokines and chemokines.5–7 TLR-mediated inflammatory cytokine production plays an important role in the activation of innate and acquired immune responses. 8 TLR4 as the receptor for LPS is the prototypical type I transmembrane receptor, characterized by an extracellular leucine-rich repeat domain and an intracellular Toll/IL-1 receptor (TIR) domain. The TIR domain is responsible for signaling and binding to TIR-containing adaptors such as Mal and MyD88. Receptor involvement via LPS enables subsequent recruitment of IL-1 receptor-associated kinases 1 and 4 (IRAK1 and IRAK4) to the TLR4/MyD88 complex and their activation. Association of IRAK1 with TNFR-associated factor 6 (TRAF-6) triggers its phosphorylation, which in turn leads to activation of NF-κB and the expression of NF-κB-induced gene products.9,10 Although a number of studies have investigated LPS-mediated signaling cascades, the exact mechanisms involved in the immune response induced by LPS remain unclear.
The phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT) signaling pathway is activated by LPS in various cell types.11,12 This pathway plays a critical regulatory role in TLR4-mediated pro- and anti-inflammatory cytokine production.12–14 Furthermore, it mediates LPS signaling, resulting in activation of the NF-κB pathway. 15 AKT activates IκB kinase (IKK), which causes the activation and subsequent phosphorylation of IκB, resulting in ubiquitin-mediated proteasomal degradation of IκB. 16 The thus liberated NF-κB translocates to the nucleus where it binds to κB motifs in the promoters of several inflammatory genes, such as TNF-α, IL-1β, iNOS, and COX-2, leading to their expression.17,18 Thus, NF-κB activation appears to play a role in cytokine expression.
In the present study, we demonstrate that, in BEAS-2B cells, the TLR4-associated kinase, IRAK1, is involved in LPS-induced TNF-α production and signaling through a direct association with MyD88. We found that IRAK1 functions a crucial component of the mechanism implicated in LPS-induced expression of TNF-α by activating the PKCα/PI3K/AKT/JNK pathway leading to activation of NF-κB.
Materials and methods
Cell culture
BEAS-2B, a human airway epithelial cell line transformed with adenovirus 12-SV40 virus hybrid, was purchased from the American Type Culture Collection. BEAS-2B cells were cultured in Dulbecco’s modified Eagle’s medium/F12 with 10% (v/v) heat-inactivated FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin at 37°C in a humidified CO2-controlled (5%) incubator.
Reagents
Fetal bovine serum (FBS), penicillin/streptomycin solution, and Dulbecco’s modified Eagle’s medium/F12 were purchased from WISENT INC (St. Bruno, QC, Canada). LPS and PDTC were obtained from Sigma (St. Louis, MI, USA). The following antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA): TLR4 monoclonal antibody, MyD88 monoclonal antibody, PKC-α polyclonal antibody, and β-actin monoclonal antibody; IRAK1 polyclonal antibody was from Abcam (Cambridge, MA, USA); JNK polyclonal antibody, p-JNK polyclonal antibody, p-PKC-α polyclonal antibody, AKT polyclonal antibody, p-AKT polyclonal antibody, p-NF-κB polyclonal antibody, Iκ-B polyclonal antibody, and p-Iκ-B monoclonal antibody were from Cell Signaling (Beverly, MA, USA); and RO320432, LY294002, SP600125, and Y-27632 from Calbiochem (San Diego, CA, USA).
Reverse transcription (RT)-PCR
cDNA was prepared from total mRNA extracted from BEAS-2B cells with TRIzol reagent, and 1 µg of RNA was reverse-transcribed using random hexamer mixed primers. The resulting cDNA was amplified by PCR. Primer sequences were as follows: TNF-α sense (5’CAGAGGGCCTGTACCTCATC3’) and antisense (5’GGAAGACCCCTCCCAGATAG3’) (PCR product, 219 bp); β-actin sense (5’AACACCCCAGCCATGTACG3’) and antisense (5’ ATGTCACGCACGATTTCCC 3’) (PCR product, 254 bp). The PCR conditions for TNF-α and β-actin were: TNF-α, denaturation at 95°C for 30 s, annealing at 60°C for 30 s, extension at 72°C for 60 s (31 cycles); human β-actin, denaturation at 95°C for 60 s, annealing at 62°C for 60 s, and extension at 72°C for 90 s (30 cycles). Amplified DNA fragments were identified on 1% agarose gel.
Western blot analysis
Cells were lysed in 20 mM Tris, pH 7.5, containing 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1% Triton X-100, 1 mM phenylmethylsulfonyl-fluoride, and 1 mM Na3VO4. Samples containing 20–30 µg protein were loaded onto SDS-polyacrylamide gels (10–12%), electrophoresed, and transferred to nitrocellulose membranes (Amersham Biosciences). After blocking with 5% dried skim milk for 1 h, the membranes were incubated with primary antibodies. The blots were further incubated with horseradish peroxidase-conjugated secondary antibody (1:2000, New England Biolabs, Beverly, MA, USA), and specific bands were detected by ECL (Ab FRONTIER, Suwon, Republic of Korea).
Small interfering RNAs
Human TLR4 (sense: 5`- GUCUAGUGGCUAAUUCCUA(dTdT) -3`, antisense: 5`- UAGGAAUUAGCCACUAGAC(dTdT)-3`) and MyD88 (sense: 5`- GAGGAAUCUGUGCUCUACU(dTdT) -3`, antisense: 5`- AGUAGAGCACAGAUUCCUC(dTdT) -3`) were purchased from Bioneer (Seoul, Republic of Korea) and IRAK1 (catalog no. L-004760-00-0005) was from Dharmacon (Denver, CO, USA). A negative control was carried out with Negative Control siRNA #2 from Ambion (Austin, TX, USA). Oligonucleotide siRNAs were transfected using siPORT™ NeoFX™ reagent (Ambion) according to the manufacturer’s instructions. At the indicated intervals following transfection, cell lysates were assayed for gene silencing by RT-PCR or western blotting.
ELISA
Cell supernatants were collected after LPS treatment and TNF-α was measured with a commercial ELISA kit (eBioscience, San Diego, CA, USA), using a standard curve in the range of 3.13–200 pg/mL. The means of triplicate ELISA values were calculated by linear regression.
Cell fractionation
To prepare nuclear extracts, the cells were washed twice with cold PBS. The cells were then transferred to microcentrifuge tubes and centrifuged at 300 ×g for 4 min at 4°C. The supernatants were discarded, and the pellets were resuspended in 400 μL cold lysis solution A (10 mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM EDTA, 1 mM DTT, 0.5 mM phenylmethylsulfonyl fluoride (PMSF)) and incubated on ice for 15 min. Then, 10 μL of 10% Nonidet P-40 (NP-40) were added, and the mixtures were vortexed briefly. Nuclei were pelleted via centrifugation at 2800 ×g for 4 min at 4°C and then resuspended in 80 μL of ice-cold lysis solution B (20 mM HEPES [pH 7.9], 0.4 MNaCl, 1 mMEDTA, 1 mMDTT, 1 mMPMSF). The mixtures were shaken vigorously for 15 min at 4°C, centrifuged at 15,000 ×g for 5 min, and then the supernatants were collected.
Statistical analysis
All experiments were repeated at least three times. Statistical comparisons were made using one-way Student’s t-test or multi-factorial ANOVA. GraphPad Prism ® (version 6; GraphPad Software, San Diego, CA, USA) was used for statistical analysis. Values of P <0.05 were considered statistically significant.
Results
LPS increases the expression and production of TNF-α in BEAS-2B cells
To determine whether LPS increased levels of TNF-α, BEAS-2B cells were treated with LPS for the indicated times. As shown in Figure 1a and b, stimulation with LPS increased TNF-α expression and production.
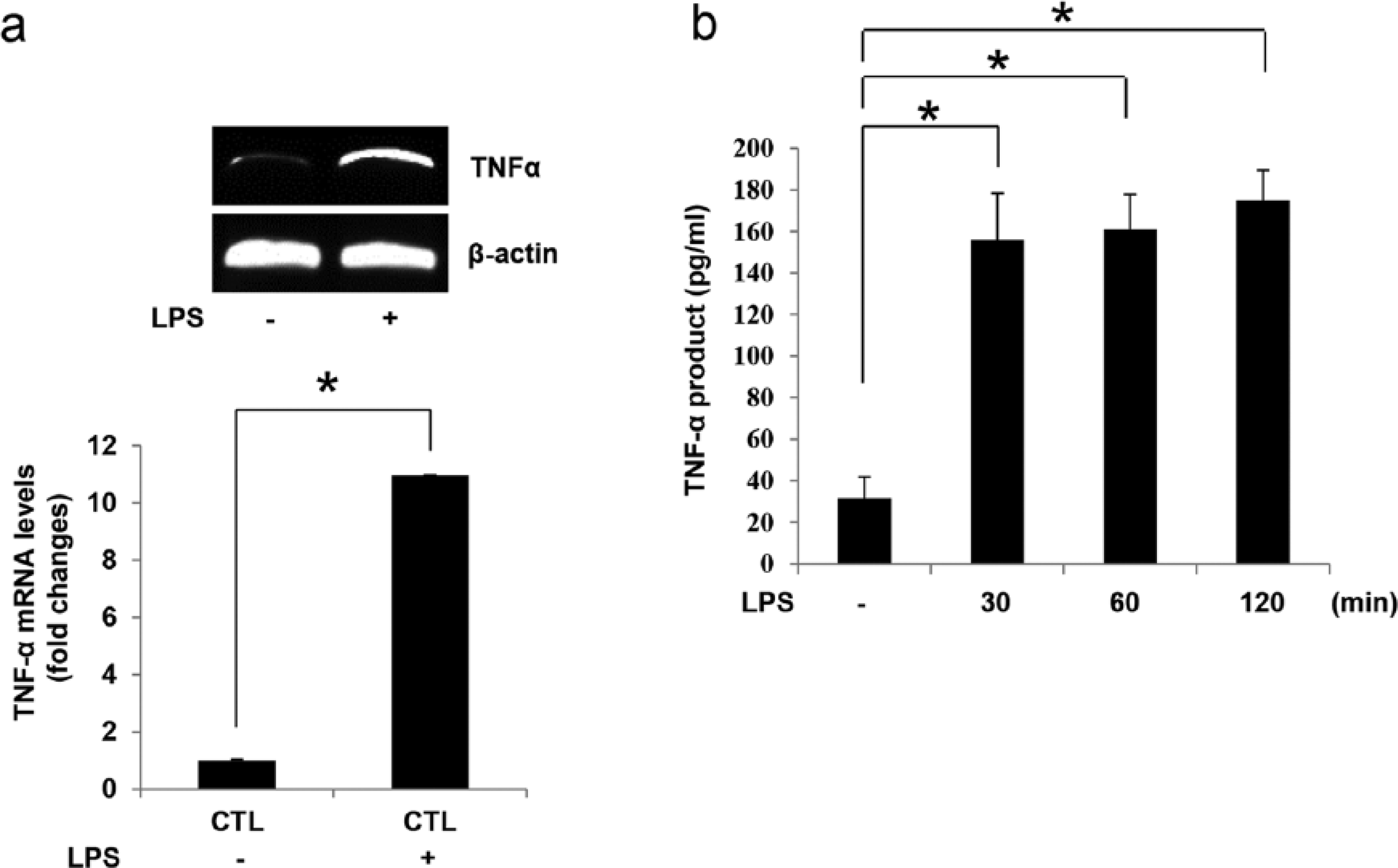
Effect of LPS on TNF-α expression and production in BEAS-2B cells. (a) BEAS-2B cells were treated with LPS (250 ng/mL) for 15 min. Total RNA was isolated using TRIzol reagent, and mRNA levels were determined by RT-PCR with primers for TNF-α or β-actin. (b) For ELISA, cells in 96-well culture plates were treated with LPS (250 ng/mL) for the indicated times. Results are the mean ± SEM in each group of samples (n = 8). *P <0.05 vs control. Bands were quantified using ImageJ software (NIH http//rsb.info.nih.gov/ij/).
Role of TLR4 in LPS-induced TNF-α expression and production
TLR4 recognizes bacterial LPS and activates inflammatory and innate immune responses, followed by intracellular signaling cascades, which ultimately lead to the production of pro-inflammatory cytokines. 19 MyD88 is a key adaptor molecule with a crucial role in the TLR-mediated signaling pathway. 20 As shown in Figure 2a, TLR4-specific siRNA completely abolished the expression of MyD88 and TNF-α induced by LPS. To confirm the involvement of TLR4 in the expression and production of TNF-α, we inhibited TLR4 with PMB, a specific TLR4 inhibitor. PMB treatment was found to completely inhibit TNF-α production (Figure 2b) indicating that TLR4 is required for the LPS-induced TNF-α expression and production in BEAS-2B cells.

Effect of TLR4 knockdown on LPS-induced TNF-α expression and production in BEAS-2B cells. (a) BEAS-2B cells were transiently transfected with 200 nM TLR4 siRNA for 48 h and stimulated with LPS (250 ng/mL) for 5 min. Cells lysates were subjected to western blotting. For RT-PCR of TNF-α, the cells were harvested and total RNA was isolated using TRIzol reagent after treatment with LPS (250 ng/mL) for 15 min. For ELISA, cells in 96-well culture plates were transfected with 200 nM TLR4 siRNA for 48 h and stimulated with LPS (250 nM) for 2 h. Results are the mean ± SEM of each group of samples (n = 8). *P <0.05 vs. LPS-treated control. (b) Cells were pretreated with PMB (50 µg) for 30 min and stimulated with LPS (250 ng/mL) for 15 min. Total RNA was isolated using TRIzol reagent, and mRNA levels were determined by RT-PCR with primers for TNF-α or β-actin. For ELISA, cells were pretreated with PMB (50 µg) for 30 min and stimulated with LPS (250 ng/mL) for 2 h. Results are the mean ± SEM of each group of samples (n=8). *P <0.05 vs. LPS-treated control. Bands were quantified using ImageJ software (NIH http//rsb.info.nih.gov/ij/).
LPS-induced TNF-α expression and production are mediated by interaction of MyD88 with IRAK1 in BEAS-2B cells
All TLRs associate directly or indirectly with MyD88, which in turn associates with Toll/IL-1R domain-containing adaptor-inducing IFN-β (TRIF) to activate downstream signaling. 21 The latter requires the association with MyD88 of some member of the IRAK family, which includes IRAK1, IRAK2, IRAK4, and IRAKM. 22 We investigated the possibility of an association between MyD88 and IRAK1. As shown in Figure 3a, MyD88 bound to IRAK1 following LPS treatment. Next, to assess the role of MyD88 and IRAK1 in TNF-α production, we downregulated MyD88 and IRAK1 by introducing MyD88- and IRAK1-specific siRNAs. As shown in Figure 3b, TNF-α expression and production were completely abolished following treatment with MyD88 siRNA, and IRAK1 siRNA had a similar effect (Figure 3c). These data suggest that the MyD88/IRAK1 complex plays an important role in the expression and production of TNF-α induced by LPS.

Interaction of MyD88 with IRAK1 is important in LPS-induced TNF-α expression and production. (a) BEAS-2B cells were treated with LPS (250 ng/mL) for 5 min. Immunoprecipitation (IP) was performed with anti-MyD88 or anti-IRAK1 antibody followed by immunoblotting for MyD88 and IRAK1. (b) BEAS-2B cells were transiently transfected with 200 nM MyD88 siRNA for 48 h and stimulated with LPS (250 ng/mL) for 5 min (for MyD88) and 15 min (for TNF-α mRNA levels). Cell lysates were subjected to western blotting. For RT-PCR of TNF-α, cells were harvested and total RNA was isolated using TRIzol reagent. For ELISA, cells in 96-well culture plates were transiently transfected with 200 nM MyD88 siRNA for 48 h and stimulated with LPS (250 ng/mL) for 2 h. Results are the mean ± SEM of each group of samples (n=8). *P <0.05 vs. LPS-treated control. (c) BEAS-2B cells were transiently transfected with 100 nM IRAK1 siRNA for 48 h and stimulated with LPS (250 ng/mL) for 5 min (for IRAK1) and 15 min (for TNF-α mRNA levels). Cells lysates were subjected to western blotting. For RT-PCR of TNF-α, cells were harvested and total RNA was isolated using TRIzol reagent. For ELISAs cells in 96-well culture plates were transiently transfected with 100 nM IRAK1 siRNA for 48 h and stimulated with LPS (250 ng/mL) for 2 h. Results are the mean ± SEM of each group of samples (n = 8). *P <0.05 vs. LPS-treated control. Bands were quantified using ImageJ software (NIH http//rsb.info.nih.gov/ij/).
PKCα is located downstream of IRAK1 in LPS-induced TNF-α expression
PKC is a central component in a signaling pathway that controls a number of cellular processes, including adaptive and innate immunity, as well as inflammation.23,24 We first confirmed that LPS stimulated the phosphorylation of PKCα (Figure 4a). Next, to determine whether the MyD88/IRAK1 complex influences LPS-induced activation of PKCα, cells were transfected with MyD88- and IRAK1-specific siRNAs. As shown in Figure 4b and c, LPS-induced PKCα phosphorylation was abolished by either MyD88- or IRAK1-specific siRNA. To confirm the role of PKCα in TNF-α expression, cells were treated with a specific PKCα inhibitor, RO320432. This completely inhibited the LPS-induced expression and production of TNF-α (Figure 4d), indicating that PKCα activation is critical in the pathway that leads to TNF-α expression induced by LPS.

PKCα is located downstream of IRAK1 in LPS-induced TNF-α expression and production. (a) BEAS-2B cells were treated with LPS (250 ng/mL) for 5 min. Cells were lysed and levels of PKCα and p-PKCα were determined by western blotting. (b) BEAS-2B cells were transiently transfected with 200 nM MyD88 siRNA or scrambled siRNA for 48 h and stimulated with LPS (250 ng/mL) for 5 min. Cells were lysed and western blots were performed for MyD88, PKCα, and p-PKCα. (c) BEAS-2B cells were transiently transfected with 100 nM IRAK1 siRNA or scrambled siRNA for 48 h and stimulated with LPS (250 ng/mL) for 5 min; cells were lysed and western blots were performed for IRAK1, PKCα, and p-PKCα. (d) Cells were pretreated with RO320432 (50 µM) for 30 min and stimulated with LPS (250 ng/mL) for 15 min. Total RNA was isolated using TRIzol reagent. RT-PCR analyses were performed using TNF-α and β-actin primers. For ELISA, cells in 96-well culture plates were pretreated with RO320432 (50 µM) for 30 min and stimulated with LPS (250 ng/mL) for 2 h. Results are the mean ± SEM in each group of samples (n = 8). *P <0.05 vs. LPS-treated control. Bands were quantified using ImageJ software (NIH http//rsb.info.nih.gov/ij/).
The PI3K/AKT pathway mediates LPS-induced TNF-α expression and production
Recent studies have demonstrated that the PI3K/AKT pathway regulates TLR4-mediated inflammatory immune responses by differentially regulating the levels of pro- and anti-inflammatory cytokines.12,13,25 Treatment of our cells with LPS led to an increase in AKT phosphorylation (Figure 5a). To test whether PKCα was involved in this effect, we inhibited it with RO320432, and found that AKT phosphorylation was also inhibited (Figure 5b). To confirm the role of AKT in TNF-α expression, cells were treated with the specific PI3K/AKT inhibitor, LY294002. This treatment completely suppressed TNF-α (Figure 5c). Together, these data suggest that the PI3K/AKT pathway is located downstream of PKCα and is required for LPS-induced TNF-α expression and production.

PI3K/AKT pathway activation is involved in LPS-induced TNF-α expression and production. (a) BEAS-2B cells were treated with LPS (250 ng/mL) for 5 min. Cells were lysed and levels of AKT and p-AKT were determined by western blotting. (b) Cells were pretreated with RO320432 (50 µM) for 30 min and stimulated with LPS (250 ng/mL) for 5 min. Lysates were subjected to western blotting. (c) Cells were pretreated with LY294002 (20 µM) for 30 min and stimulated with LPS (250 ng/mL) for 15 min. Total RNA was isolated using TRIzol reagent. RT-PCR analyses were performed using TNF-α and β-actin primers. For ELISA, cells in 96-well culture plates were pretreated with LY294002 (20 µM) for 30 min and stimulated with LPS (250 ng/mL) for 2 h. Results are the mean ± SEM of each group of samples (n = 8). *P <0.05 vs. LPS-treated control. Bands were quantified using ImageJ software (NIH http//rsb.info.nih.gov/ij/).
JNK activation is important in LPS-induced TNF-α expression and production
MAP kinases are critical controllers of cellular responses to inflammatory substances such as NO, COX-2, and pro-inflammatory cytokines.26,27 JNK is a MAP kinase responsible for mediating inflammation in response to extracellular stimuli, and it controls gene expression by phosphorylating transcription factors.28,29 As shown in Figure 6a, LPS treatment induced JNK phosphorylation. To determine whether the PI3K/AKT pathway was involved, cells were pretreated with LY294002. We found that the resulting inhibition of PI3K/AKT completely suppressed the phosphorylation of JNK (Figure 6b). To further elucidate the role of JNK in TNF-α expression, cells were pretreated with SP600125, a specific JNK inhibitor. This inhibition completely suppressed the expression and production of TNF-α (Figure 6c). Thus JNK appears to play an important role in the LPS-induced expression and production of TNF-α, and acts downstream of the PI3K/AKT pathway.
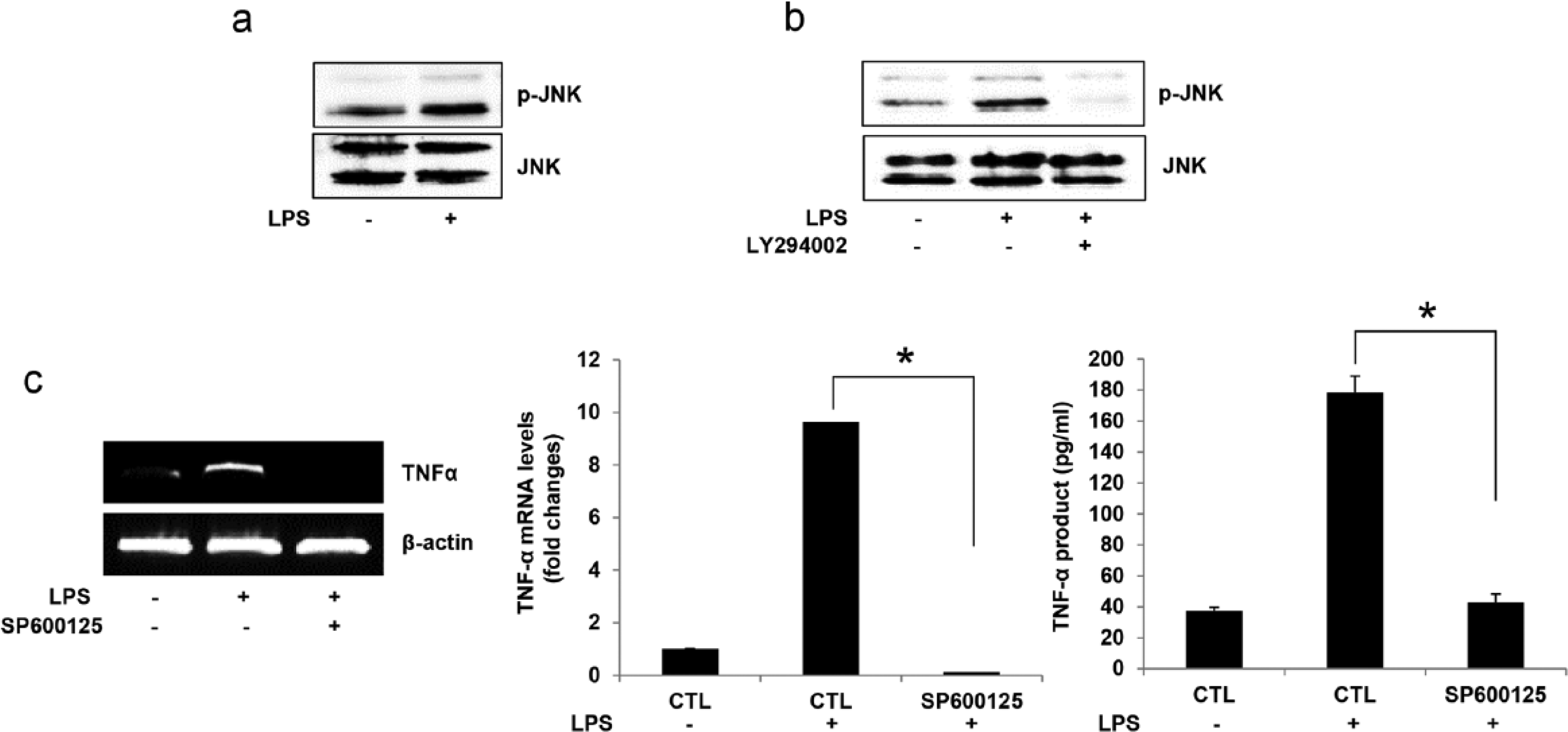
JNK activation is important for LPS-induced TNF-α expression and production. (a) BEAS-2B cells were treated with LPS (250 ng/mL) for 5 min. Cells were lysed and levels of JNK and p-JNK were determined by western blotting. (b) Cells were pretreated with LY294002 (20 µM) for 30 min and stimulated with LPS (250 ng/mL) for 5 min. Cells lysates were subjected to western blotting. (c) Cells were pretreated with SP600125 (50 μM) for 30 min and stimulated with LPS (250 ng/mL) for 15 min. Total RNA was isolated using TRIzol reagent. RT-PCR analyses were performed using TNF-α and β-actin primers. For ELISA, cells in 96-well culture plates were pretreated with SP600125 (50 μM) for 30 min and stimulated with LPS (250 ng/mL) for 2 h. Results are the mean ± SEM of each group of samples (n = 8). *P <0.05 vs. LPS-treated control. Bands were quantified using ImageJ software (NIH http//rsb.info.nih.gov/ij/).
JNK activates NF-κB which acts as a transcription factor for TNF-α expression
NF-κB plays an important role in regulating inflammatory and immune responses to extracellular stimuli.30,31 Some studies have provided evidence that activation of TLR4 by LPS results in the activation of MAPK and NF-κB, and eventually in the release of inflammatory cytokines such as IL-6, IL-1β, and TNF-α.32,33 Therefore, we examined whether NF-κB activation might be responsible for the production of TNF-α induced by LPS. As expected, treatment with LPS increased the frequency of events associated with NF-κB activation such as phosphorylation of p65 and IκBα, and degradation of IκBα (Figure 7a). Next, to see whether JNK was involved in NF-κB activation, cells were pretreated with SP600125, a specific JNK inhibitor. As shown in Figure 7b, JNK inhibition blocked LPS-induced NF-κB activation. Furthermore, LPS increased the translocation of NF-κB p65 from cytosol to nucleus, and this increase was completely inhibited by pretreatment with SP600125. Similarly, treatment with an NF-κB inhibitor, PDTC, completely inhibited TNF-α expression and production (Figure 7c). These results suggest that NF-κB is an important transcription factor for TNF-α, and is activated by JNK.

Effect of NF-κB on LPS-induced TNF-α expression and production in BEAS-2B cells. (a) BEAS-2B cells were treated with LPS (250 ng/mL) for 5 min. Cells were lysed and levels of p-p65, IκBα, and p-IκBα were determined by western blotting. (b) Cells were pretreated with SP600125 (50 μM) for 30 min and stimulated with LPS (250 ng/mL) for 5 min. Cells lysates were subjected to western blotting (upper panel). Nuclear and cytosol fractions (lower panel) were subjected to 10% SDS–PAGE and analyzed with p65 antibody. (c) Cells were pretreated with PDTC (100 μM) for 1 h and stimulated with LPS (250 ng/mL) for 15 min. Total RNA was isolated using TRIzol reagent. RT-PCR analyses were performed using TNF-α and β-actin primers. For ELISA, cells in 96-well culture plates were pretreated with PDTC (100 μM) for 1 h and stimulated with LPS (250 ng/mL) for 2 h. Results are the mean ± SEM of each group of samples (n = 8). *P <0.05 vs. LPS-treated control. Bands were quantified using ImageJ software (NIH http//rsb.info.nih.gov/ij/).
Discussion
LPS, a strong immune stimulant, promotes the expression and production of inflammatory cytokines in various cell types, leading to an acute inflammatory response to pathogens. 34 Regulation of LPS-mediated cytokine production is a key step in immunity-related diseases. In response to LPS, peritoneal macrophages secrete inflammatory cytokines including IL-1β, IL-6, TNF-α, and NO, which play a critical role in macrophage activation and are associated with acute and chronic inflammation.35–37 TNF-α is not only an inflammatory mediator, but also stimulates the release of other inflammatory mediators and causes the acute phase reaction. 38 However, the molecular mechanism by which LPS induces cytokines in human bronchial epithelial cells is not fully understood. The aim of our study was to provide insight into the TLR4-mediated signaling pathway involved in the production of TNF-α in response to LPS in human bronchial epithelial cells.
When activated by their agonists, TLRs trigger various signaling cascades that lead to cytokine production and initiation of an adaptive immune response. 39 TLR expression is known to increase in a plethora of inflammatory disorders.40,41 Generally LPS activates cells through TLR4 with the help of the accessory proteins CD14 and MyD88. TLRs activate two types of downstream signaling pathways: MyD88-dependent and MyD88-independent. MyD88 is an immediate and widespread downstream adaptor molecule, recruited by activated TLRs through their Toll-IL-1 receptor domain. MyD88, in turn, recruits IRAK1, and induces phosphorylation. IRAK1 associates with TRAF6, which leads to activation of the IKK complex, and subsequently to that of the NF-κB transcription factor. The activity of the MyD88-dependent signaling pathways leads to the induction of inflammatory cytokines including TNF-α, IL-6, and IL-1β. 42 Importantly, our data demonstrate that activation of TLR4 induced by LPS leads to a similar recruitment of IRAK1, and this pathway appears to be involved in TNF-α expression and production.
There is evidence that PKC is one of the main signal transduction systems involved in the inflammatory response.43,44 Alongside PKC, PI3K/AKT is an important pathway involved in LPS-mediated signaling. 45 For this reason, we posed the question whether PKCα could be involved in an LPS-induced PI3K/AKT pathway. We found that pretreatment with a specific PKC inhibitor completely blocked the LPS-induced activation of AKT and production of TNF-α. As expected, the PI3K/AKT inhibitor attenuated the production of TNF-α, suggesting that LPS-induced activation of PI3K/AKT in BEAS-2B cells was dependent on PKCα activation.
To elucidate this mechanism further, we investigated the downstream molecules regulated by the LPS-induced PI3K/AKT pathway. Previous studies have demonstrated that the activation of MAPK cascades could proceed through a variety of upstream factors that include PK3K/AKT. 46 MAPKs play an important role in cytokine production. 47 At the same time, stimulation of various cell types with LPS has been shown to induce the phosphorylation and activation of ERK, p38 MAPK, and JNK.48,49 which in turn signals the expression of pro-inflammatory cytokines. Some research groups in particular have suggested that the JNK signaling pathway controls the inflammatory response. However, those workers did not examine the relationship between the PI3K/AKT pathway and JNK, which regulates TNF-α production in the immune response. In the present study, we found that LPS stimulated JNK activation, which was then completely abolished by inhibition of the PI3K/AKT pathway. Furthermore, a JNK inhibitor attenuated the production of TNF-α. Therefore, it is highly likely that the PI3K/AKT pathway acts as an upstream signaling pathway in LPS-induced JNK activation. This observation also leads to the assumption that JNK activation by LPS is important for TNF-α expression and production.
NF-κB is a well-known master regulator of inflammatory responses. 50 When not activated it is localized in the cytosol as a homodimer, or as a heterodimer with IκB. After simulation with LPS, NF-κB is activated by IκB phosphorylation, and regulates the expression of a network of inflammatory cytokines such as TNF-α, IL-6, and IL-1β.32,51 Therefore, activation of NF-κB plays an important role in inflammation through its ability to induce the transcription of pro-inflammatory genes.32,52 Previous studies reported that LPS activated NF-κB to cause the release of inflammatory cytokines (such as TNF-α) through a TLR4-dependent inflammatory pathway.53,54 Here, we found that inhibition of NF-κB blocked TNF-α expression and production, suggesting that activated NF-κB plays an important role in the pathway that leads to TNF-α expression and production in response to LPS.
In conclusion, our experiments provide new evidence that LPS-induced TNF-α expression is mediated by the PKCα/PI3K/AKT/JNK/NF-κB signaling pathway, which is dependent on MyD88/IRAK1 (Figure 8).

A model of the signaling pathway involved in LPS-induced TNF-α expression in BEAS-2B cells. Our model suggests that LPS-induced expression of TNF-α occurred through TLR4/MyD88/IRAK1/PKCα/PI3K/AKT/JNK/NF-κB pathway.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was supported by the National Research Foundation of Korea (NRF), funded by the Mid-Career Researcher Program (NRF-2010-0026844), the Korea government (MEST) (NRF-2013R1A2A2A03067895), and Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2013R1A1A2061420).