
Abstract
The innate immune system in mammals include pattern recognition receptors (PRRs), which initiate immune responses to microbial infection via several mechanisms. These PRRs include cell surface Toll-like receptors (TLRs) and cytosolic Nod-like receptors (NLRs) that recognizes extracellular and intracellular danger signals respectively. NLRs are poised to respond specifically to pathogens that access the host cell cytosol. The molecular mechanisms by which NLRs are activated to form inflammasomes and exert downstream inflammatory responses remain poorly understood. Additionally, very little is known about the regulation of cytosolic pathogen sensory NLR family members, except for NLRP3. Recently a deubiquitinase known as STAMBP has been implicated as a regulator of NLRP7 inflammasome assembly. We have investigated the role of STAMBP in regulation of other inflammasome components and its broader role in inflammation using genetic removal of STAMBP protein from cells using CRISPR/Cas9 gene editing and challenging these gene edited cells with an inflammatory stimuli. Our study demonstrated that STAMBP has a critical role in inflammation both in the context of NLR pathway, through NLRP stabilization and TLR pathway, through JNK signaling and downstream cytokine production. The findings indicate that STAMBP has a wider role in inflammation than previously thought to be the case.
Introduction
Inflammation is the body’s immediate response against injury and infection. Wounds and any damage to tissue would not heal without an inflammatory response. Inflammation is a short-term reaction which is switched off once the damage is repaired. However, inflammation can become harmful when it continues long after the original response was initiated. While it’s a relatively complicated process, inflammation becomes “chronic” if it fails to shut off when it should and therefore, becomes uncontrolled. 1
Pattern recognition receptors (PRRs) play a crucial role during inflammation by acting as sensors to detect pathogens and molecules typical for the pathogens (“danger signals”). There are two classes of PRRs. These are the Toll-like receptors (TLRs) that sense extracellular danger signals and Nod like receptors (NLRs) that sense the intracellular danger signals. 2 Activation of TLRs at the cell surface triggers downstream signal transduction and gene transcription through nuclear factor-κB (NF-κB) and c-Jun N-terminal kinase (JNK) proteins. Activated NLRs induces the assembly and activation of the inflammasome in the cytosol, a crucial multiprotein complex for cellular inflammatory response. 3 The end result of the PRR signal transduction is the production of pro-inflammatory cytokines and a lytic type of cell death known as pyroptosis in the host cell (Supplemental Figure S1). 4 Among PRR families, Nod-like receptors (NLR) have attracted significant attention for their crucial function in modulating the organization of inflammasomes. 5 Several members including NLRP1, NLRP2, NLRP3, NLRC4, NLRP6, NLRP7, and NLRP12 can form multimeric inflammasome complexes. NLRP3 containing inflammasome is one of the best-studied inflammasomes but other inflammasomes, including those containing NLRP1 and NLRP7 have been identified yet understudied. 6
In a 2017, Lys63-specific deubiquitinase STAM-binding protein (STAMBP) was identified to be an interacting partner for NLRP7. This study showed that NLRP7 is constitutively degraded by the proteasome in the absence of PRR activation. However, when the PRRs are activated by danger signals, STAMBP removes Lys63 ubiquitin chains from NLRP7 resulting in its stabilization and accumulation in cells. This was shown using the human monocytic cell line, THP-1. 7
STAMBP, is a gene encoding the deubiquitinating (DUB) isopeptidase STAMBP (STAM-binding protein)/AMSH (Associated Molecule with the SH3 domain of STAM). It is known to play a key role in cell surface receptor-mediated endocytosis and sorting. The lack of STAMBP and hence impaired endosomal sorting is associated with the intracellular accumulation of ubiquitinated proteins, resulting in microcephaly-capillary malformation syndrome in knockout mice.8−10 Apart from these reports, little is known about the role of STAMBP. Recently, STAMBP was implicated in NLRP7 mediated inflammation. 7 Additionally, other studies have suggested STAMBP as an inflammation serum marker in late pregnancy for assessing risk of postpartum depression 11 and chronic pain,12,13 hinting a role for it in inflammation. According to the Human Protein Atlas data, STAMBP is expressed at moderate levels in all malignant tissues with high levels in a few cases of pancreatic, gastric, and prostate cancers.
In this study, we explored whether STAMBP has an prominent role in stabilization of NLRPs besides NLRP7 and whether it has a more broader role in inflammation in addition to preventing NLRP7 degradation.
Methods
Cell culture
Human LIM1215 colon cancer cell and THP-1 macrophage cell lines were cultured in RPMI 1640 medium and HEK293T cells in DMEM (GIBCO Life Technologies) at 37°C in a humidified incubator with 5% CO2. All media were supplemented with 10% (v/v) fetal calf serum (FCS), 100 U/ml penicillin/streptomycin (GIBCO Life Technologies). Cell were stimulated in culture using Nigercin sodium salt (Sigma-Aldrich) and Lipopolysaccharide (LPS) (Sigma-Aldrich).
Western Blotting
The cell lysates were prepared in RIPA lysis buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, % NP40) with added protease inhibitor cocktail and PhosSTOP phosphatase inhibitor cocktail (Roche). The protein concentration was determined using the BCA Protein Assay Kit (Pierce). Proteins were size fractionated by gel electrophoresis on 12% Tris-Glycine gels in MES buffer. See Blueplus2 molecular weight ladder (Life Technologies) was use as a reference for determining the protein size. Following electrophoresis the protein bands were transferred onto a nitrocellulose membrane (Life Technologies) using the iBlot membrane transfer system and blocked using 5% skim milk in PBS-T. The following antibodies were diluted in 5% skim milk in phosphate buffered saline-tween (PBS-T) buffer to probe the membranes. Anti-β-actin, Anti-phospho-JNK (Thr183/Tyr185), Anti-JNK, Anti-STAMBP, Anti-NLRP1, Anti-GAPDH, Anti-IκB and Anti-P-p65 antibodies (Cell Signaling Technologies). Anti-NLRP7 and Anti-Pro-Caspase (Santa Cruz Biotechnology). Following probing with primary antibody the membranes were probed with IRdye conjugated secondary antibody (1:100,000) for 1 h at room temperature. The antibodies used were IRDye 800CW goat anti-mouse and goat anti-rabbit IRDye 680RD from LI-COR. After washing the membrane, the fluorescent signals were visualized with ODYSSEY CLx (LI-COR®) machine.
Immunoprecipitation experiments
Pierce™ Classic Magnetic IP/CO-IP Kit (Thermo scientific) was used according to manufacturer’s protocol for immunoprecipitation. Cells at 70% confluence, were washed with 1X PBS (137 mM NaCl, 2.7 mM KCl, 1.8 mM KH2PO4, 10 mM Na2HPO4) prior to lysing with lysis/wash buffer. Cell lysates (1 mg) were incubated with 10 μg of anti-NLRP7/anti-STAMBP overnight at 4°C. Then the immune complex was added to pre-washed protein A/G magnetic beads and incubated at RT for 2 h with continuous mixing. The beads were collected using Thermo Scientific™ MagJET separation rack and the unbound fractions and wash fractions were stored for further analysis. The beads containing the bound proteins were then resuspended in 50 μL of 4X SDS loading dye and quantified using SYPRO® Ruby fluorescent dye stain (Molecular probes). Samples were stored in −80°C until further analysis.
Generation of CRISPR mediated gene knockout cells
To generate Cas9 expressing cell lines, HEK293T cells were co-transfected with the following plasmids; pFH1tUTG-Cas9, pCMV ∂R8.2, and pVSV using Lipofectamine LTX transfection reagent (Invitrogen, Life Technologies). Forty-eight hours post transfection viral supernatant was harvested, filtered, combined with Polybrene (Sigma-Aldrich) and used to infect target cells by spin infection at 2500 g at 37°C. Twenty-four hours post infection cells were expanded and several passaged later sorted for mCherry positive cells expressing Cas9 using BDFACS Aria. Guide RNA was designed using CRISPR design portal at crisp.mit.edu. The guide RNA sequences used are as follows. The first four bases indicate the overhangs that are added to facilitate the cloning.
Forward guide RNA: 5′ TCCC CCA GAG CGG AAG TAC CGA CG 3′
Reverse guide RNA: 5′ AAAC CGT CGG TAC TTC CGC TCT GG 3′
Guide RNA were annealed and cloned into pFGH1tUTG vector and transfected into Hek293T cells along with pCMV ∂R8.2 and pVSV plasmids. The 48 h viral supernatant used to infect Cas9 cells. Twenty-four hours post-infection cells were expanded and 1 μg/mL doxycycline was added to the media for four consecutive days. Single cells were sorted into 96 well plates based on mCherry and FITC double positivity. Once clones were visible in 96 well plates they were expanded further and lysed for Western blot analysis to screen for gene knockout cells which lack the targeted protein expression.
Preparation of conditioned media
Preparation of conditioned media (CM) was carried out as previously described. 14 For preparing CM, LIM1215 cells were seeded in 100 mm diameter culture dishes (three dishes per cell line) in the presence of 7 mL of culture media. After cell density reached 70% to 80% confluence, the cells were washed with PBS. About 7 mL of fresh media with or without 10 μg/mL LPS was added on to cells. Twenty-four hours later CM was collected, centrifuged 500 g to remove floating cells followed by another centrifugation step at 2000 g for 20 min to get rid of apoptotic debris. The CM was concentrated 10 times using Amicon Ultra-15 centrifugal device with 3000 Da nominal molecular weight limit (Millipore) at 4000 g. The resulting concentrated CM was stored at −80°C until further analysis.
Cytokine measurements in conditioned media
IL-8, IL-6, and TNF-α in the concentrated conditioned media was measured using the cytometric bead array assay kit (BD Biosciences) as described previously. 15 Briefly, the bead cocktail was prepared by mixing capture bead and detection reagent for cytokines. To a 96-well V-bottom culture plate (Greiner Bio-One), bead cocktail was incubated with an equal volume of cytokine standards or culture supernatants in the dark for 3 h at room temperature. Following two washes at 200 g for 5 min, the samples were reconstituted in wash buffer and detected using a BD FACSCanto II Flow Cytometer and BD FACSDiva Software v6.1.1, acquiring at least 300 events per capture bead. The data were analyzed using FCAPArray v.3 software (BD Biosciences).
Statistical analysis
Statistical analysis was performed with Prism5 (GraphPad) and Microsoft Office Excel. All data shown are representative of results obtained from experiments conducted three times. The results were analyzed by T-tests. Denotes statistical significance (*p < 0.05). ImageJ software were used for Western blot image analysis.
Results
NLRP7 protein levels and its interaction with STAMBP is enhanced during inflammation
Under pathological conditions such as Crohn’s disease and ulcerative colitis, the mucosa of the intestine is infiltrated by inflammatory cells. The infiltrated immune cells can crosstalk with the epithelial cell layer of the intestine and regulate its function. Both the immune cells and epithelial cells can mount pro-inflammatory responses through the inflammasome formation. The activated immune cells secrete many pro-inflammatory cytokines such as TNF-α, which can bind receptors on the epithelial cells to further promote inflammation. Using the colon epithelial cells LIM1215 stimulated with 40 ng/mL TNF-α, we were able to demonstrate the stabilization of NLRP7 protein (Figure 1(a)). The TNF-α treatment resulted in activation of NF-κB signaling as seen by the degradation of inhibitor of kB (IkB) at 30 min post stimulation. It is possible that NLRP7 is one of the NF-κB transcriptional targets in addition to procaspase-1. However, this hypothesis is yet to be established, although it is known that NF-κB transcriptionally targets NLRP3. 16 These data also suggest that NLRP7 has a role in inflammation in colon epithelial cells.

NLRP7 protein levels and its interaction with STAMBP during inflammation. (a) After stimulation of LIM1215 cells with TNF-α and the downstream NF-κB signaling and NLRP7 stabilization was analyzed by Western blot. GAPDH is used as a loading control for Western blots. The numbers under each Western blot panel indicates the band intensities normalized to the band intensity of the loading control. (b) Immunoprecipitation was carried out using anti-STAMBP and anti-NLRP7 antibodies for stimulated and unstimulated LIM1215 cell lysates and the proteins that were pulled-down were used for Western blot analysis.
Using an immunoprecipitation experiment, Bednash et al., has previously reported STAMBP can interact with NLRP7 in THP-1 macrophage cells and that STAMBP has a role in stabilization of NLRP7 during inflammation. 7 Inflammation is a key process in disease pathologies such as colorectal cancer from ulcerative colitis as the colon epithelium is constantly in contact with inflammatory stimuli such as bacteria. 17 Therefore, we used LIM1215 colorectal carcinoma cells and stimulated them with 10 μg/mL LPS (for TLR signaling activation) and 10 μM Nigericin (for NLR signaling activation) to initiate inflammation. The whole cell lysates were extracted for immunoprecipitation experiments using anti-STAMBP or anti-NLRP7 antibodies. Equal volumes of the immunoprecipitated samples were subjected to Western blotting. Immunoprecipitation was also carried out with unstimulated LIM1215 cell lysates to determine the interaction between NLRP7 and STAMBP at basal levels. More STAMPB protein was pulled-down in the stimulated samples by anti-NLRP7 antibody, suggesting that, under inflammatory conditions, these proteins interaction is enhanced (Figure 1(b)). This phenomenon maybe also be due to increased stabilized NLRP7 by STAMBP under stimulation. Our data show that this interaction is not limited to immune cells but also occurs in non-immune cells. Similarly, in the reverse immunoprecipitation where anti-STAMBP antibodies are used, along with STAMBP, procaspase-1, whose role in inflammation is well-established, was also pulled-down. These immunoprecipitation data suggested that STAMBP interacts with not only NLRP7, but also other inflammasome components such as pro-caspase1. 18
STAMBP is required for stabilization of NLRP1
It has been previously shown that STAMBP is required for NLRP7 stabilization. 7 To test if this is also the case for other NLRPs, STAMBP was genetically deleted from LIM1215 cells using CRISPR gene-editing (Figure 2(a)). The overall levels of ubiquitinated proteins were increased in STAMBP knockout cells (Supplemental Figure S2). Changes in inflammasome components were observed in STAMBP knockout cells both under stimulated (with 10 μg/mL LPS and 10 μM Nigericin) and unstimulated conditions (Figures 2(b) and Supplemental Figure S3(a)). STAMBP knockout cells had less NLRP1 levels both at basal (0 h) and stimulated time points (Figure 2(b)). However, even in the STAMBP knockouts, an extremely faint band for NLRP1 could be seen at 2 h post stimulation which disappeared by 4 h time point indicating some upregulation at translational level by upon stimulation before rapid degradation. These data suggest that similar to NLRP7, NLRP1 is constitutively degraded as STAMBP is no longer available to remove its Lys63 ubiquitination in order to prevent degradation. The STAMBP knockout cells had surprisingly high basal pro-caspase 1 levels and low proIL-1β (a pro-cytokine that is produced by NF-κB signaling) levels (Supplemental Figure S3(a)). This observation requires further investigation. On the contrary, the stability of the more widely studied NLRP3 protein was not affected by knockout of STAMBP (Supplemental Figure S3(b)). This hints at a non-redundant role between NLRP3 and other NLRPs, particularly NLRP1 and NLRP7 investigated in this study. NLRP3 regulation appears to be STAMBP independent.
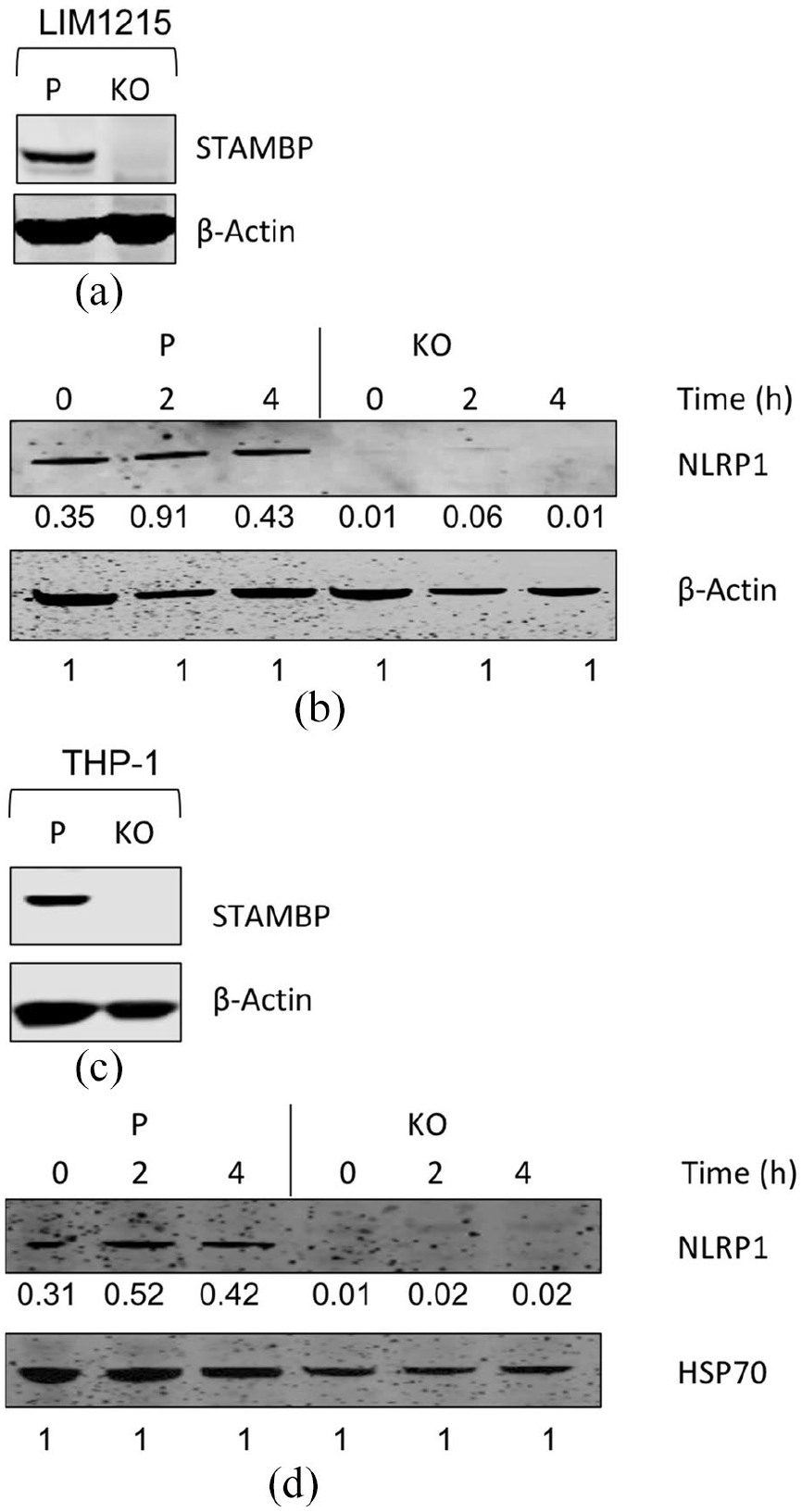
Western blot analysis for parental and STAMBP knockout LIM1215 Cas9 and THP-1 Cas9 cells under basal and inflammatory conditions. (a) STAMBP protein expression in parental and CRIPSR mediated gene knockout (KO) LIM1215 Cas9 cells. (b) Parental and STAMBP knockout LIM1215 Cas9 cells were treated with LPS and Nigericin for indicated time points and immunoprobed for proteins of interest. The numbers under each Western blot panel indicates the band intensities normalized to the band intensity of the loading control. (c) STAMBP protein expression in parental and CRIPSR mediated gene knockout (KO) THP-1 Cas9 cells. (d) Parental and STAMBP knockout THP-1 Cas9 cells were treated with LPS and Nigericin for indicated time points and immunoprobed for proteins of interest. The numbers under each Western blot panel indicates the band intensities normalized to the band intensity of the loading control. β-Actin or HSP70 is used as a loading control for Western blots.
The inflammation in the intestine is due to the interplay between both the intestinal epithelial cells and the immune system. Therefore, it is important to study the role of NLRPs and their interactors in the context of immune cells as well. Hence, the STAMBP knockouts were also made in THP-1 cells using CRISPR based gene-editing. The successful knockouts were confirmed by the lack of STAMBP protein expression in Western blot analysis (Figure 2(c)). Similarly, to that observed in STAMBP knockout LIM1215 intestinal epithelial cells, these knockouts also showed changes in NLRP1 (Figure 2(d)).
JNK signaling is impaired during inflammation in STAMBP knockouts cells
To investigate if STAMBP has a role in inflammation beside NLRP stabilization (through NLRs), we looked into the JNK and NF-κB signaling pathways (through TLR activation with 10 μg/mL LPS alone) and their downstream cytokines production using the parental and STAMBP gene knockout cells (refer to Supplemental Figure S1 for clarification). JNK signaling was altered in STAMBP knockout cells compared to parental Cas9 cells. Phosphorylation of JNK (P-JNK) is a marker for activated JNK signaling. JNK signaling was not activated upon stimulation of STAMBP knockouts (i.e. P-JNK does not increase in STAMBP knockouts as seen with parental at 6 h) and even the basal levels of P-JNK was abolished in the knockout cells at 24 h (Figure 3(a)). However, NF-κB signaling was not altered in STAMBP knockout cells compared to parental cells upon stimulation (Figure 3(b)). As a result of reduced JNK signaling, consequent downstream IL-6 and TNF-α production was also drastically reduced in STAMBP knockout cells compared to parental cells when cells were stimulated (Figure 3(c) and (d)). In the conditioned media from unstimulated cells, IL-6 and TNF-α could be barely detected as the basal levels are very low. IL-8 levels were unaffected in the knockouts as it is controlled by NF-κB signaling (Figure 3(e)).

JNK signaling and NF-κB signaling during inflammation in STAMBP knockouts cells. (a and b) Parental and STAMBP knockout LIM1215 Cas9 cells were stimulated with LPS. Whole cell lysates were prepared at different time points after stimulation and immunoprobed for proteins of interest. β-Actin or GAPDH is used as a loading control for Western blots. The numbers under each Western blot panel indicates the band intensities normalized to the band intensity of the loading control. (c and e) For cytokine assay, 24 h after the stimulation the conditioned media was collected and concentrated 10 times. The concentrated media was used for the cytokine detection by cytometric bead array assay kit. *Denotes statistical significance (*p < 0.05).
Discussion
Our study demonstrated that STAMBP has a critical role in inflammation both in the context of NLR pathway (through NLRP stabilization) and TLR pathway (through JNK signaling and downstream cytokine production). Though previously only reported for NLRP7, 7 our data have extended the STAMBP-mediated stabilization to NLPR1, a member of the large and complex NLR protein family. Although the stimuli and molecular mechanisms underlying the activation of most NLRs remain poorly understood, it is already clear that these stimuli and mechanisms differ considerably across NLR family members. 19 Striking differences are apparent even in the comparison of just two NLRs, the NLRP1 and NLRP3 inflammasomes. NLRP3 is one of the most studied NLRs and is known to be stimulated by a wide variety of agonists, but no single unifying molecular model appears to account for NLRP3 activation. We observed that its mode of activation differs from both NLRP1 and NLRP7 in terms of regulation by STAMBP. Thus, further studies of the NLR family are required to gain insight into how their regulation is associated with STAMBP. Interestingly, we also observed that the role of STAMBP is not limited to the NLR pathway, but rather has a novel function in TLR-mediated JNK signaling and downstream cytokine production.
There are several limitations in this study that needs to be addressed. The role of STAMBP in the formation of inflammasome complex itself needs to be further validated besides the NLRP1 stabilization at protein levels. It would be interesting to see if the NLRP1 inflammasomes in the cells would be reduced in STAMBP knockout cells upon stimulation. Also, to confirm whether the stabilization of NLRP1 by STAMBP is due to the deubiquitination and hence reduced proteosomal degradation the stimulation of STAMBP knockout cells should be carried out in the presence of a proteosomal inhibitor. It would be also interesting to perform a two step immunoprecipitation for NLRP1 first using a anti NLRP1 antibody and then a UBA pull down (for ubiquitinated proteins) with lysates from stimulated and non-stimulated parental and STAMBP knockout cells. This will clarify whether STAMBP mediated removal of ubiquitinin is indeed the reason for NLRP1 stabilization in the presence of an inflammatory stimuli. Inspite of these points that needs further addressing, this data provides the first indication of a role for STAMBP in NLRP1 and JNK signaling regulation (Supplemental Figure S4). It is likely that STAMBP might even have a wider role in inflammation and potential studies using models of inflammation in STAMBP knockout mice might help shed light on its multiple roles. Given that lack of STAMBP promotes a phenotypic switch from a pro-inflammatory state to anti-inflammatory, it might be a potential target for controlling undesirable inflammation.
Conclusion
The new findings of this study are that (1) in addition to NLRP7, the NLPR1 abundance is also modulated by STAMBP. (2) STAMBP regulates the TLR-mediated JNK signaling and downstream cytokine production. Overall, this study demonstrate a broader role for deubiquitinating isopeptidase STAMBP in inflammation than previously thought to be the case.
Footnotes
Acknowledgements
We would like to acknowledge Marco J Herold Lab at the Walter and Eliza Hall Institute, Melbourne, Australia for providing pFH1tUTG-Cas9, pCMV ∂R8.2 and pVSV plasmids.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a Victorian Cancer Agency Early Career Seed Grant (grant number ECSG15018.)