
Abstract
Introduction
Nucleotide-binding oligomerization domain-like receptor family, pyrin domain containing 3 (NLRP3), an intracellular pattern recognition receptor, recognizes various pathogen-associated molecular pattern and/or damage-associated molecular pattern molecules to constitute inflammasome that act as an interleukin (IL)-1β processing platform. Injected insulin is reported to induce focal amyloidosis and the formation of subcutaneous lumps called insulin balls, but the formation of subcutaneous lumps and the underlying cytotoxic mechanism has not been elucidated.
Methods
Amyloid formation was evaluated by thioflavin T spectroscopic assay and scanning electron microscopy. Binding between insulin amyloid fibrils and NLRP3 was evaluated by immunoprecipitation followed by native polyacrylamide gel electrophoresis. Inflammasome activation was evaluated by immunofluorescence speck formation called “ASC speck” and Western blotting. IL-1β secretion in culture supernatants of peripheral blood mononuclear cells was evaluated by enzyme-linked immunosorbent assay. Cytotoxicity was measured by lactate dehydrogenase release assay.
Results
Insulin amyloid fibrils interact directly with NLRP3, resulting in NLRP3 inflammasome activation and pyroptotic cell death.
Conclusion
Insulin ball formation and cytotoxicity may be associated with NLRP3 inflammasome activation followed by pyroptotic cell death.
Introduction
Amyloidosis is the tissue deposition of soluble and/or insoluble aggregates of amyloid proteins characterized by rigid and unbranched structures. 1 Systemic amyloidosis is the deposition of amyloid proteins in various tissues leading to multiple organ failure due to cytotoxicity. 2 Injected insulin is reported to lead to focal amyloidosis and the formation of subcutaneous lumps called insulin balls. 3 Mori et al. 4 recently reported insulin-derived amyloid could be cytotoxic in focal injected subcutaneous tissues in patients with diabetes, but the cytotoxicity mechanism was not assessed.
Nucleotide-binding oligomerization domain-like receptor family, pyrin domain containing 3 (NLRP3), an intracellular pattern recognition receptor (PRR), recognizes various pathogen-associated molecular pattern molecules and damage-associated molecular pattern molecules to constitute a large complex “inflammasome” that act as an interleukin (IL)-1 processing platform. 5 We previously reported that islet amyloid polypeptide (IAPP)/amylin and β-amyloid (Aβ) directly interacts with NLRP3 to activate NLRP3 inflammasome.6,7 In the present study, we demonstrated that insulin amyloid fibrils interact directly, clarifying the association between insulin-derived amyloidosis and the NLRP3 inflammasome.
Methods
Reagents
Human insulin was purchased from FUJIFILM Wako Chemicals (Tokyo, Japan). Anti-insulin rabbit monoclonal antibody (mAb; ab181547) and anti-insulin guinea pig polyclonal antibody (pAb; ab7842) were purchased from Abcam (Cambridge, UK). Anti-Aβ mouse mAb (6E10) was purchased from Bioregend (San Diego, CA, USA). Peroxidase-conjugated goat anti-mouse immunoglobulin G (IgG; NBP1-75172), anti-rabbit IgG (NBP1-75339), and anti-guinea pig IgG (106-035-006) were purchased from Jackson Immuno Research (West Grove, PA, USA). A Chemi-Lumi One Markers Kit was purchased from NACALAI TESQUE INC. (Kyoto, Japan). Thioflavin T (ThT) was obtained from Sigma-Aldrich (St. Louis, MO, USA).
Preparation of insulin amyloid
Insulin was dissolved in 100 mM NaCl and 25 mM HCl at a final protein concentration of 2 mg/mL (344 µM). The concentration of insulin was determined by the bicinchoninic acid assay using a Pierce BCA Protein Assay Kit (Pierce). For fibril formation, insulin monomers (344 μM) were incubated at 65°C for 5 h. Samples incubated for shorter time (2.5 h) were used for comparison as an intermediate sample.
Thioflavin T spectroscopic assay
Insulin samples (5 μM) were mixed with ThT solution (1 μM) in phosphate-buffered saline, and ThT fluorescence was measured using a microplate reader at an excitation wavelength of 450 nm and an emission wavelength of 490 nm.
Native polyacrylamide gel electrophoresis (PAGE) and Western blotting
Insulin samples (2 mg/mL) were mixed with native-PAGE sample buffer at a final protein concentration of 0.87 mg/mL and subjected to 10% native-PAGE. Precision Plus Protein Dual Color Standards (Bio-Rad, Hercules, CA, USA) were used as molecular mass markers. Following transfer to a nitrocellulose membrane, the membrane was blocked with 5% skim milk in Tris-buffered saline (TBS) for 24 h at room temperature. The membrane was then incubated with anti-insulin rabbit mAb (ab181547, 1:1000) for 24 h at room temperature. After washing with TBS, the membrane was incubated with anti-rabbit IgG secondary antibody (1:2000) for 3 h at room temperature. After washing with TBS, proteins were visualized using ECL Select Western Blotting Detection Reagent (GE Healthcare, Chalfont St. Giles, UK). Luminescence was detected by an Image Quant LAS4010 instrument (GE Healthcare).
Immunoprecipitation
NLRP3-PYD and NLRP3-LRR proteins were synthesized using wheat germ cell-free system as described previously. 6 Insulin (2.0 μg) and 1 μg of C-terminal biotinylated pyrin domain (exons 1−2) of NLRP3 (NLRP3-PYD-biotin) or C-terminal biotinylated leucine-rich repeat (LRR) domain (exons 4−9) of NLRP3 (NLRP3-LRR-biotin) were lysed in 300 μL of NP-40 buffer comprising 1% Nonidet P-40, 142.5 mM KCl, 5 mM MgCl2, 10 mM HEPES pH 7.6, 0.2 mM phenylmethylsulfonyl fluoride (PMSF), and 1 mM EDTA. Anti-insulin rabbit mAb (ab181547; 2 μg) and 20 μL of protein A-conjugated agarose beads (Invitrogen, Waltham, MT, USA) were added and incubated for 3 h at 4°C. Samples were subjected to sodium dodecyl sulfate (SDS)-PAGE and immunoblotting. Detection on blotting membranes was performed using horseradish peroxidase (HRP)-conjugated streptavidin and anti-insulin guinea pig pAb (ab7842) followed by secondary anti-guinea pig IgG (106-035-006).
Next, 1.6 μg of Aβ and 1 μg of C-terminal FLAG-tagged pyrin domain (exons 1−2) of NLRP3 (NLRP3-PYD-FLAG) or C-terminal FLAG-tagged LRR domain (exons 4−9) of NLRP3 (NLRP3-LRR-FLAG) were lysed in 300 μL of NP-40 buffer. As described above, 2 μg of anti-Aβ mAb (6E10) and 20 μL of protein A-conjugated agarose beads (Invitrogen) were incubated for 3 h at 4°C. Samples were subjected to SDS-PAGE and immunoblotting. Detection on blotting membranes was performed using anti-FLAG M2 monoclonal antibody (Sigma-Aldrich) and anti-Aβ mAb (6E10) followed by secondary anti-mouse IgG (NBP1-75172).
Scanning electron microscopy
Insulin samples (5 μM) were placed on a silicon wafer and air-dried. Samples were observed at an acceleration voltage of 15 kV using a field emission scanning electron microscope (FE-SEM, JSM7001FA, JEOL, Tokyo, Japan).
Inflammasome activation assay
ASC-speck is a marker for activation of inflammasome. We counted the ratio of ASC-specks in cells using immunofluorescence microscopy to evaluate inflammasome activation as previously reported. 8 Human peripheral blood mononuclear cells (PBMCs) were incubated with insulin monomers, intermediates, fibrils, or vehicle (buffer) for 8 h. Cells in each well were fixed on glass slides and incubated with anti-ASC mouse mAb, 9 followed by Alexa Fluor 488 AffiniPure F(ab')2 fragment goat anti-mouse IgG (H+L) antibody (115-546-146, Jackson ImmunoResearch, West Grove, PA, USA) and monitoring by immunofluorescence microscopy to evaluate ASC-speck.
Detection of IL-1β expression and activation
Expression of IL-1β and activation of IL-1β was detected by Western blotting and enzyme-linked immunosorbent assay. Each PBMC lysate was subjected to SDS-PAGE followed by Western blotting. Pro-IL-1β and cleavage of IL-1β were evaluated by Western blotting using an anti-IL-1β (D3U3E) rabbit mAb #12703 (Cell Signaling Technology, Danvers, MA, USA) and an anti-cleaved-IL-1β (Asp116) (D3A3Z) rabbit mAb #83186 (Cell Signaling Technology), respectively. IL-1β concentrations in PBMCs culture supernatants of each well were measured by OptEIA Set for human IL-1β (BD Biosciences, San Jose, CA, USA) according to the manufacturer’s instructions.
Cytotoxicity assay
Cytotoxicity was assessed by measurement of the release of lactate dehydrogenase (LDH) activity using CytoTox96 (Promega, Madison, WI, USA) according to the manufacturer’s instructions.
Statistical analysis
All results are presented as the mean ± standard deviation (SD) of data from three independent experiments and compared by Mann–Whitney U-tests. A p-value <0.05 was considered statistically significant.
Ethics approval and guidelines for human experiments
PBMCs from the healthy volunteer with written informed consent were approved by the Human Research Ethical Committees of Ehime University (No.1712006).
Results
Insulin alters the biochemical and morphological properties of amyloid fibrils
Formation of insulin amyloid fibrils was analyzed by ThT assay, and the results revealed an increase in ThT fluorescence intensity, indicating the formation of insulin amyloid fibrils (Figure 1(a)). The ThT intensity of the intermediate sample incubated for shorter time was lower than that of the fibrils. The formation of insulin amyloid fibrils was further confirmed by native-PAGE followed by Coomassie Brilliant Blue (CBB) staining and Western blotting (WB; Figure 1(b)). The blotting membrane revealed intermediates of polymerization and fibrils from monomers to fibrils (Figure 1(b)). Insoluble species stacked in the top of the gel were observed for the fibrils, supporting the formation of insoluble aggregates. Similar results were obtained using CBB staining (Figure 1(b)). SEM analysis also confirmed differences between insulin monomers, intermediates, and fibrils (Figure 1(c)). Notably, shorter fibrils were formed in the intermediate sample, consistent with the obtained results (Figure 2(a) and (b)). Insulin oligomerization and morphological changes. (a) ThT assay results showing the formation of insulin amyloid under oligomerization conditions at 65°C for 0 h (monomers), 2.5 (intermediates), or 5 h (fibrils). (b) Insulin monomers (2 mg/mL) were incubated under oligomerization conditions at 65°C for 0 h (monomers), 2.5 h (intermediates), or 5 h (fibrils) and subjected to 10% native polyacrylamide gel electrophoresis followed by Western blotting (WB) and Coomassie Brilliant Blue (CBB) staining. (c) Scanning electron microscopy (SEM) images of insulin samples under oligomerization conditions at 65°C for 0 h (monomers), 2.5 h (intermediates), or 5 h (fibrils). Scale bars = 1 μm. Insulin fibrils interact with NLRP3-LRR. (a) Schematic representation of the full-length NLRP3 structure depicting exon components and truncated forms. The pyrin domain (PYD) is indicated by black boxes, the nucleotide-binding oligomerization domain (NOD) is indicated by a light gray box, and leucine-rich repeats (LRR) are indicated by striped boxes. Amino acid sequence numbers are also indicated. (b) FLAG-tagged truncated forms of NLRP3 proteins and Aβ fibrils were precipitated by anti-Aβ mouse mAb conjugated with protein A and detected by WB with anti-FLAG mouse mAb or anti-Aβ mouse mAb. (c) Biotinylated truncated forms of NLRP3 proteins and insulin fibrils were precipitated with anti-insulin rabbit mAb conjugated with protein A followed by WB with streptavidin or anti-insulin guinea pig pAb.

Formation of insulin amyloid fibrils induces interaction between insulin and NLRP3
Full-length NLRP3 (NLRP3-FL) and its truncated forms NLRP3-PYD and NLRP3-LRR are shown schematically in Figure 2(a). The LRR domain of NLRP3 (NLRP3-LRR) is involved in the recognition of pathogens, and NLRP3-FL resides in the inactive state through loose interactions between PYD and LRR in the absence of upstream signals. 10 When NLRP3-LRR-FLAG was coprecipitated with IAPP/amylin, antibody light chain was evident as a non-specific band. 6 Therefore, C-terminal biotinylated proteins NLRP3-PYD-biotin and NLRP3-LRR-biotin instead of Flag-tagged proteins were subjected to immunoprecipitation assays. As expected, when NLRP3-LRR-FLAG was coprecipitated with Aβ fibrils (controls), the presence of the light chain of the antibody was negligible (asterisk, Figure 2(b)).
Using wheat germ cell-free system-specific expression plasmids, NLRP3-LRR-biotin and NLRP3-PYD-biotin were successfully produced (Figure 2(c)). Next, we investigated whether insulin fibrils interacted with NLRP3-LRR using immunoprecipitation. The results showed that when precipitated with anti-insulin antibody, NLRP3-LRR-biotin was coprecipitated with insulin fibrils, whereas NLRP3-PYD-biotin was not, suggesting that insulin fibrils can interact with the LRR domain (Figure 2(c)).
Insulin fibrils activate NLRP3 inflammasome
To investigate whether insulin fibrils activate NLRP3 inflammasome leading to IL-1β secretion, human PBMCs were incubated with insulin monomers, intermediates, fibrils, or vehicle with or without LPS for 8 h. Pro-IL-1β expressions and cleavages were evaluated with SDS-PAGE followed by WB (Figure 3(a) left panel, center panel, and right panel). When incubated with 10 μM insulin fibrils, there was no pro-IL-1β band or significant IL-1β secretion in culture supernatants without LPS (Figure 3(a) left panel and Figure 3(b) upper panel). Notably, even without LPS, LDH secretions (Figure 3(c)) and the percentage of ASC-specks upon incubation with fibrils were significantly higher than that with monomers, intermediates, or vehicle (Figure 3(d) left panels and Figure 3(e) upper panel). When incubated with 10 μM insulin fibrils with 0.01 ng/mL and 0.1 ng/mL LPS, pro-IL-1β bands were appeared (Figure 3(a) left panel). A cleaved-IL-1β band was evident only when incubated with 10 μM fibrils together with 0.1 ng/mL LPS (Figure 3(a) center panel). Consistent with this, IL-1β secretion in culture supernatants upon incubated with 10 μM fibrils together with 0.1 ng/mL LPS was significantly higher than that with monomers, intermediates, or vehicle (Figure 3(b) lower panel). The percentage of ASC-specks upon incubated with 10 μM fibrils together with 0.1 ng/mL LPS was higher than that with monomers, intermediates, or vehicle (Figure 3(d) right panels and Figure 3(e) lower panel). Insulin fibrils activate inflammasome. (a) Western blotting analysis of pro-IL-1β production (left panel) and IL-1β cleavage (center panel; the same blotting membrane shown in the upper panel was re-hybridized with a mAb specific for cleaved-IL-1β) using PBMCs incubated with 10 μM insulin amyloid fibrils together with indicated amounts of LPS. The same gel was stained with Coomassie Brilliant Blue (CBB, right panel). (b) IL-1β concentrations in the PBMCs culture supernatant of each well measured by ELISA. *p-value < 0.05 (Mann–Whitney U-test). (c) Lactate dehydrogenase (LDH) activity in the PBMCs culture supernatant of each well (A490). *p-value < 0.05 (Mann–Whitney U-test). (d) Immunofluorescence microscopy evaluation of ASC-speck formation. Scale bars = 10 μm. (e) Percentage of ASC specks (%) in PBMCs without (upper panel) or with (lower panel) 0.1 ng/mL LPS. *p-value < 0.05 (Mann–Whitney U-test).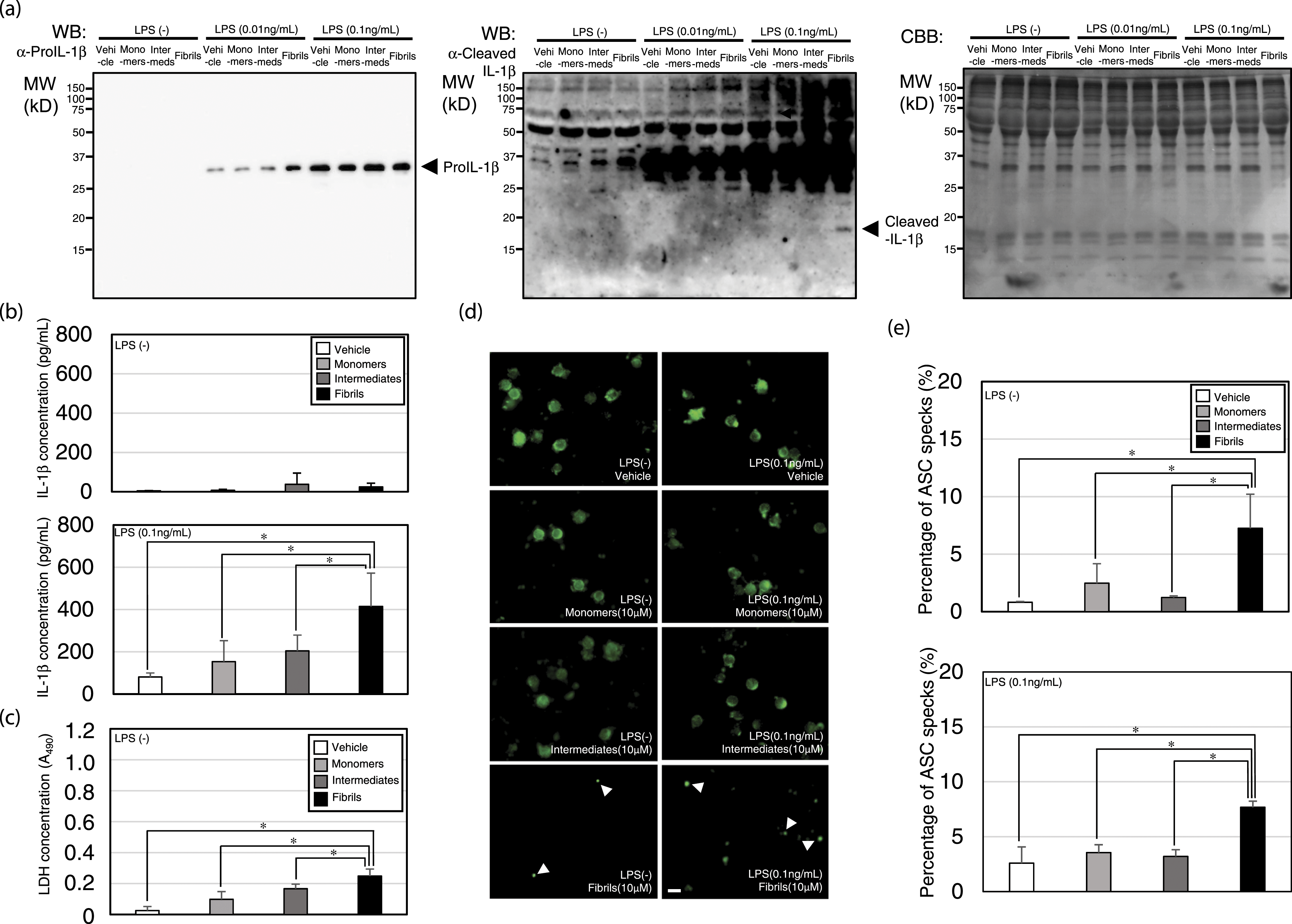
Discussion
Insulin-derived amyloidosis occurs in subcutaneous injected tissue in patients with diabetes and is characterized by insulin balls. 4 It was reported that some insulin balls are cytotoxic, while others are not, implying amyloid polymorphism and structural differences between high- and low-toxicity species. 5
In terms of general pathology, the formation of insulin balls is involved in amyloidosis leading to inflammation and fibrosis, 11 and the cytotoxicity of insulin appears to be linked to amyloid pathology. Formation of insulin balls is a form of reversible cell injury, and cytotoxicity is an example of irreversible cell injury. Both types of cell injury were induced by DAMPs, which can be sensed by PRR such as Toll-like receptors and Nod-like receptors (NLRs). 12 Previous work reported that NLRP3 inflammasome activation is closely linked to inflammation via the IL-1β activation pathway and through pyroptotic cell death. 13
In the present study, we successfully prepared insulin fibrils (Figure 1(a)–(c)). During their preparation, we are aware that insulin is reported to be mostly exist as dimeric rather than monomeric at pH 1.6 (Figure 1(b)), and monomers and dimers can be separated under high temperature condition for nucleation. 14 Consistently, slight monomer and strong dimer bands were observed in “Monomers” lane, and monomer band was much more intense below the dimers band in the “Fibrils” lane (Figure 1(b)).
The insulin fibrils and Aβ fibrils can interact directly with the NLRP3-LRR and this leads to inflammasome activation (Figure 2). Master et al. reported that IAPP/amylin triggered the NLRP3 inflammasome and generated mature IL-1β, and Halle et al. reported that the NLRP3 inflammasome is a sensor of Aβ.15,16 We previously demonstrated that amyloid proteins of IAPP/amylin and Aβ directly interact with NLRP3 via the LRR domain, depending on their amyloid properties during the transition to fibrils and thereby activating NLRP3 inflammasomes.7,10 Therefore, insulin amyloid fibrils may activate the NLRP3 inflammasome in a similar context. This inflammasome activation induced pyroptotic cell death in PBMCs, but IL-1β was released from PBMCs only when pro-IL-1β was expressed in PBMCs (Figure 3). This may explain why insulin balls do not always appear in patients with diabetes.
IL-1β is reported to be closely related to the mesenchymal transition and fibrosis. 17 Since insulin balls consist of fibrocollagenous nodes at the injection site, their pathology may be associated with pro-IL-1β expression via chronic inflammation. 18
Conclusion
Insulin ball formation may be associated with NLRP3 inflammasome activation and systemic chronic inflammation which induces pro-IL-1β expression.
Footnotes
Acknowledgments
Scanning electron microscopy was carried out using a field emission scanning electron microscope at the Division of Material Science of the Advanced Research Support Center (ADRES), Ehime University.
Author contributions
WM, NK, and JM conceived and devised the study. WM, NK, AN, TZ, and JM were responsible for data acquisition. WM, NK, AN, and JM analyzed the data. All authors contributed to data interpretation. WM and JM drafted the manuscript. All authors critically reviewed and edited the manuscript. JM led this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Grants-in-Aid for Scientific Research through JSPS KAKENHI grant numbers 20H03719 (JM), 20H01085 (NK), and 19H02527 (TZ) from the Ministry of Education, Culture, Sports, Science and Technology, Japan (JM), and the Research Unit for Advanced Nano-Bioanalysis from Ehime University (TZ and JM).