
Abstract
Background
Meningoencephalitis can occur in myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease (MOGAD).
Objective
To assess the clinical and radiologic features of MOG-IgG meningoencephalitis.
Methods
Multicenter retrospective cases series of MOG-IgG meningoencephalitis and literature review of MOG-IgG-positive patients with the clinical syndrome of meningoencephalitis.
Results
Ninety MOG-IgG-positive patients were identified from three academic medicine centers. 8/90 (8.9%) patients presented with a clinical syndrome of meningoencephalitis (age: 4–57 years; 5/8 male; 4/8 Caucasian; MOG-IgG titers 1:20–1:1000), which was the initial presentation in 7/8 patients. Symptoms included headaches (n = 8/8), encephalopathy (n = 7/8), seizures (n = 4/8), meningismus (n = 3/8), and aphasia (n = 3/8). Cerebrospinal fluid (CSF) pleocytosis was present in 7/8 patients (12–1745 cells/mm3) and frequently neutrophilic (>25%; n = 4/6). Magnetic resonance imaging (MRI) was notable for leptomeningeal enhancement with (n = 5/8) or without cortical edema (n = 1/8), focal dural enhancement (n = 1/8), and leptomeningeal loss of FLAIR suppression (n = 1/8). 7/8 patients sustained a relapsing disease course. Literature review identified 150 additional cases with MOG-IgG meningoencephalitis (initial attack in 86.7%) with median age of 20 (1–67) years, Asian (90.4%) and male (54.1%) predilection, and CSF pleocytosis in 87.9% of patients (82 [0–887] cells/cm3; ≥100 cells in 44.3%), which was frequently (51.7%) neutrophilic.
Conclusions
MOG-IgG meningoencephalitis may represent the initial presentation of MOGAD with neutrophilic pleocytosis in CSF and meningo-cortical involvement on MRI.
Keywords
Introduction
Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease (MOGAD) can present with a broad spectrum of different presentations including optic neuritis, myelitis, brainstem demyelination, and acute disseminated encephalomyelitis (ADEM).1,2 Furthermore, it can present as a clinical syndrome of meningoencephalitis with meningo-cortical manifestations (i.e. cerebral cortical encephalitis [CCE] and FLAIR-hyperintense Lesions in Anti-MOG-associated Encephalitis with Seizures [FLAMES]) as well as predominantly meningeal presentations (i.e. FLAIR-variable Unilateral Enhancement of the Leptomeninges [FUEL] and MOG antibody-associated Aseptic Meningitis [MOGAM]). 3 The objective of this study is to assess the clinical and radiologic features of MOG-IgG meningoencephalitis.
Methods
Case series
This is a multicenter retrospective review of all MOG-IgG-positive cases with the clinical syndrome of meningoencephalitis at the University of Florida (UF), Baylor College of Medicine (BCM), and the University of California San Diego (UCSD). The study was approved by the UF, UCSD, and BCM institutional review boards.
MOG-IgG-positive cases were identified by review of the respective institution's cases of ICD10 diagnosis code G36 as well as the individual institution's neuromyelitis optica spectrum disorder/MOGAD clinical databases. Only MOG-IgG-positive patients with ≥1 clinical presentation of meningoencephalitis were included. The clinical syndrome of meningoencephalitis was defined as presence of ≥2 cardinal symptoms (fever, nuchal rigidity, altered mental status, and headache). 4 Subsequently, all included cases were reviewed if they fulfilled the international MOGAD panel proposed criteria. 1
For collection of pertinent clinical data, electronic medical records (EHR; Epic Systems Corporation, Verona, WI, USA) were reviewed. Collected data included patient demographics, the initial syndrome of presentation, cerebrospinal fluid (CSF) results, the disease course (monophasic or relapsing), antibody titers including serum and CSF MOG-IgG, aquaporin-4 (AQP4) IgG, N-methyl-D-aspartate receptor (NMDAR) IgG, and glial fibrillary acidic protein (GFAP) IgG as well as utilized immunotherapies. A clinical relapse was defined as a new clinical attack occurring >30 days following the onset of a previous attack. Response to acute treatment was rated on the 7-point Clinical Global Impression-Global Change (CGI-C) scale (1 = very much improved, 2 = much improved, 3 = minimally improved, 4 = no change, 5 = minimally worse, 6 = much worse, 7 = very much worse). Data analysis of cases series was completed using descriptive statistics.
Literature review
A literature search was conducted via PubMed database filtering for articles in English language, published up until 12/31/2023, and utilizing terms: “unilateral cerebral cortical encephalitis,” “FLAMES,” “FUEL,” “MOG encephalitis,” “MOG meningoencephalitis,” “anti-MOG antibody-associated encephalitis,” and “cerebral cortical encephalitis.” We included all studies that reported unique patients with the clinical syndrome of meningoencephalitis. According to the preferred reporting items for systematic review and meta-analysis (PRISMA) guidelines, the literature search was conducted by two reviewers (AE and TR). Discussions were held until final agreement was reached. A detailed PRISMA flow chart is shown in Figure 1. From the published data, we extracted demographic data (age, sex, race/ethnicity), the clinical presentation (timepoint of the meningoencephalitis [initial presentation versus relapse] and the occurrence of seizures), magnetic resonance imaging (MRI) features (reported FLAIR hyperintensities, edema, and contrast enhancement), CSF findings (cell count, protein), serum MOG-IgG titers, available serum and CSF autoimmune encephalitis antibodies including NMDAR-IgG and GFAP-IgG, utilized immunotherapies including intravenous methylprednisolone, intravenous immunoglobulins, and corticosteroid tapers, the treatment outcome, and the occurrence of relapses. Data analysis of literature review subjects was completed using descriptive statistics.

Flow charts. (a) Flowchart of the retrospective chart review depicting identified patients from each participating center that met the criteria of a clinical syndrome of meningoencephalitis. (b) PRISMA 2020 flowchart for study selection and use in literature review. UF = University of Florida, BCM = Baylor College of Medicine, UCSD = University of California San Diego, PRISMA = Preferred reporting items for systematic review and meta-analysis.
Results
Of 90 total (UF: n = 54; BCM: n = 14; UCSD: n = 32) MOG-IgG-positive cases, 8 (8.9%) patients (age range: 4–57 years; 5/8 male; 4/8 Caucasian, 2/8 Asian, 1/9 African American, and 1/8 Hispanic) presented with a clinical syndrome of meningoencephalitis with headaches (n = 8/8), encephalopathy (n = 7/8), seizures (n = 4/8), nausea/vomiting (n = 3/8), meningismus (n = 3/8), and aphasia (n = 3/8) (Figure 1, Table 1). Serum MOG-titers were assessed with a live cell-based assay (Mayo Clinic Laboratories, Rochester, MN, USA) in all patients and resulted as 1:20 (n = 1), 1:40 (n = 2), 1:100 (n = 3), and 1:1000 (n = 2). CSF MOG-titers were only available in 1 patient and positive with an immunoglobulin-binding-index of 4.61 and a titer of 1:32 (Mayo Clinic Laboratories, Rochester, MN, USA). All cases fulfilled the 2023 international MOGAD panel criteria 1 at the initial presentation. MOG-IgG meningoencephalitis represented the initial presentation of the disorder in 7/8 patients and the first relapse in 1 patient. Alternate diagnoses including Multiple Sclerosis and other neuroinflammatory diseases were ruled out with standard testing methods. Serum AQP4 antibodies were negative in all patients (live cell-based assay; Mayo Clinic Laboratories, Rochester, MN, USA).
Patient characteristics, diagnostic work-up, and treatment, relapsing versus monophasic disease course.
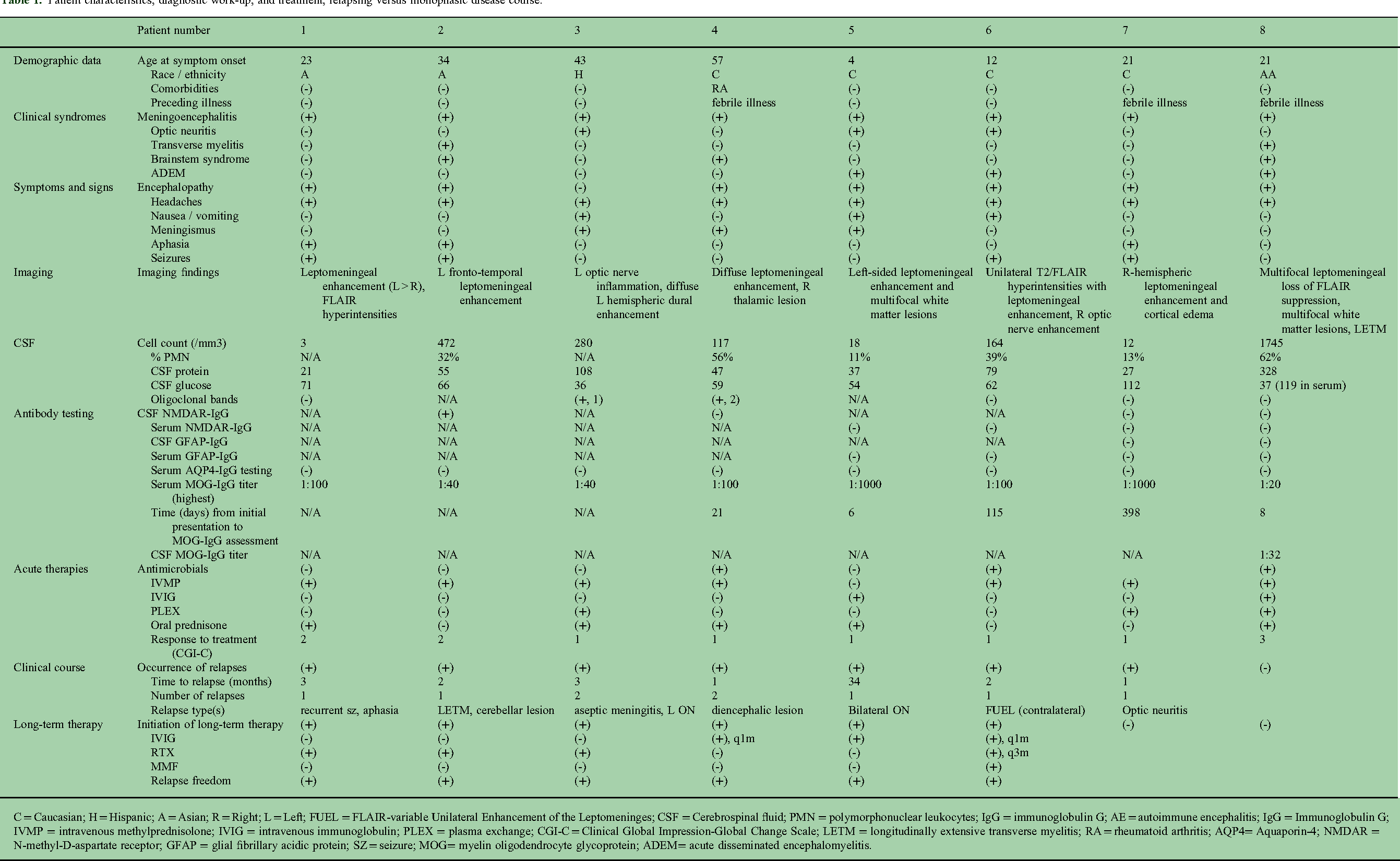
C = Caucasian; H = Hispanic; A = Asian; R = Right; L = Left; FUEL = FLAIR-variable Unilateral Enhancement of the Leptomeninges; CSF = Cerebrospinal fluid; PMN = polymorphonuclear leukocytes; IgG = immunoglobulin G; AE = autoimmune encephalitis; IgG = Immunoglobulin G; IVMP = intravenous methylprednisolone; IVIG = intravenous immunoglobulin; PLEX = plasma exchange; CGI-C = Clinical Global Impression-Global Change Scale; LETM = longitudinally extensive transverse myelitis; RA = rheumatoid arthritis; AQP4= Aquaporin-4; NMDAR = N-methyl-D-aspartate receptor; GFAP = glial fibrillary acidic protein; SZ = seizure; MOG= myelin oligodendrocyte glycoprotein; ADEM= acute disseminated encephalomyelitis.
CSF studies were available in all patients and notable for a pleocytosis in 7/8 patients (12–1745 cells/mm3) which was frequently (n = 4/6) neutrophilic (defined as >25% polymorphonuclear neutrophils [PMN] 5 ). Low numbers (1 and 2 bands) of CSF-restricted oligoclonal bands (OCBs) were present in 2/8 patients. Serum and CSF NMDAR-IgG testing was available in four patients each with CSF NMDAR-IgG antibody positivity in one patient and otherwise negative results. Serum and CSF GFAP-IgG testing was available in four and two patients, respectively, and negative in all cases (Table 1).
MRI revealed unilateral leptomeningeal enhancement with cortical edema (n = 5/8), unilateral focal intense dural enhancement (n = 1/8), leptomeningeal enhancement without cortical edema (n = 1/8), and bilateral focal leptomeningeal loss of FLAIR suppression (n = 1/8) (Figure 2).

Magnetic resonance imaging of patient cases. Axial (a, c, e, g, I, k, n) and coronal (m) T2/FLAIR and axial contrast-enhanced T1-weighted sequences (b, d, f, h, j, l, o). (a, b)
Acute treatment of MOG-IgG meningoencephalitis consisted of intravenous methylprednisolone in 7/8 patients and 7/8 patients sustained a favorable outcome with reports of “much” to “very much improved” on the CGI-C scale.
7/8 patients had a relapsing disease course including recurrent meningoencephalitis (n = 2) and subsequent exacerbations with longitudinally extensive transverse myelitis (n = 1), optic neuritis (n = 3), and diencephalic involvement (n = 1). A monophasic disease course was observed in one patient (follow-up: 11 months) under a prolonged prednisone taper. Six patients were subsequently started on disease-modifying therapy with immunoglobulin (n = 3/6) and rituximab (n = 3/6) and remain relapse free to date.
Literature review
We reviewed a total of 91 articles, excluding 23 articles due to clinical presentation being other than meningoencephalitis (Figure 1). We included 68 articles consisting of 150 unique patients with MOG-IgG meningoencephalitis (Table 2, supplementary file 1).6–75 Median age was 20 years [1–67 years, IQR 11–30 years], 80/148 (54.1%) were male (2 with no documented sex), and 103/114 (90.4%; 36 with no documented race/ethnicity) were of Asian race. MOG-IgG meningoencephalitis was the initial attack of MOGAD in 130/150 (86.7%) patients. 122/150 (81.3%) of patients met 2023 international MOGAD panel criteria at initial presentation and 126/150 (84.0%) fulfilled panel criteria at follow-up. 1 Besides the clinical syndrome of meningoencephalitis, clinical presentations included seizures in 95/150 (63.3%), optic neuritis/ocular symptoms (n = 40/149 [26.8%]), and less commonly transverse myelitis and brainstem syndromes seen in 22/144(15.3%) and 12/114 (10.5%), respectively. Imaging characteristics revealed unilateral FLAIR hyperintensities in 74/144 (51.4%) and bilateral FLAIR hyperintensities in 60/144 (41.7%). Contrast enhancement (leptomeningeal and/or parenchymal) was reported in 62.8% (86/137). CSF studies often showed elevated cell count (n = 123/140 [87.9%] with median [range] of 82 [0–887] cells/cm3) with 44.3% (62/140) having 100 cells or greater. Notably, 15/29 (51.7%) cases where differential was reported demonstrated a neutrophilic pleocytosis (defined as >25% PMNs 5 ). CSF protein was elevated in 70/129 (54.3%) patients and CSF-restricted OCBs were absent in the majority of patients (59/69 [85.5%]). Serum MOG-IgG titers were available in 82/150 patients with a titer ≥1:100 in 54/82 (65.9%). CSF NMDAR-IgG co-positivity was reported in 13/118 (11.0%) patients and CSF GFAP-IgG co-positivity in 2 patients. Acute treatments consisted of combination therapies including intravenous methylprednisolone (n = 133/150), oral corticosteroids (n = 87/150), and intravenous immunoglobulin (n = 47/147). 87/148 (58.7%) patients were documented to have achieved “full recovery.” However, relapses were reported in 55/145 (37.9%) patients with 48/145 (33.1%) occurring ≥1 month from the prior episode (fulfilling the criteria of a new clinical attack 1 ). 35/60 (58.3%) of relapses occurred within 1 year of diagnosis, most commonly within 1 month but ranging from 1 month to >10 years (23 relapses did not have documented timeframe of occurrence). Long-term immunotherapy was initiated in 56/148 (37.8%) of patients and consisted mostly of intravenous immunoglobulin, mycophenolate mofetil, rituximab, and azathioprine. 43.9% (65/148) of patients were placed on an oral steroid taper.
Aggregate table of literature review of MOGAD meningoencephalitis via PubMed database filtering for articles in English language and utilizing terms: “unilateral cerebral cortical encephalitis,” “FLAMES,” “FUEL,” “MOG encephalitis,” “MOG meningoencephalitis.” A total of 68 articles were included consisting of 150 unique patients.6–75 Aggregate data is presented as median (range) and number (percentage) of patients. Detailed data from all included studies is presented in supplementary file 1.

MOG= myelin oligodendrocyte glycoprotein; MOGAD= myelin oligodendrocyte glycoprotein antibody-associated disease; FLAMES= FLAIR-hyperintense Lesions in Anti-MOG-associated Encephalitis with Seizures; FUEL= FLAIR-variable Unilateral Enhancement of the Leptomeninges; CSF= Cerebrospinal fluid; PMN= polymorphonuclear neutrophils.
Discussion
In this multicenter retrospective cohort of 90 MOG-IgG-positive patients, we identified that meningoencephalitis is a rare clinical presentation (8.9%) that often occurs as the initial clinical attack, may predict a relapsing disease course, and frequently occurs in the presence of neutrophilic pleocytosis in CSF, with meningeal and cortical MRI findings.
Comparing our cohort to the 150 cases identified in our literature review, they were similar with regard to slight male predilection, elevated CSF cell count with neutrophilic pleocytosis, largely negative OCB status, and the majority of relapses occurring within 1 year of diagnosis. In our cohort, 2/8 (25.0%) of patients were Asian which is more than expected for the patient population of our centers and may be in concordance with the Asian predominance seen in the literature review (103/114 [90.4%;] of patients) which is in contrast to MOGAD in general where no obvious racial preponderance has been observed so far. 76 However, given that this predominance is derived from an aggregate of case reports, it could represent publication bias and will need to be further evaluated by population-based studies. There appears to be a subset of MOG-IgG meningoencephalitis patients with CSF NMDAR-IgG co-positivity, as seen in one patient from our cohort. Our cohort differed with regard to relapse rates (87.5% in our cohort compared to 37.9% in previously published cases).
The case of profound (>1000 cells/mm3) neutrophilic pleocytosis with association hypoglycorrhachia (P8) strikingly demonstrates that MOG-IgG meningoencephalitis may even mimic a bacterial infection. Accordingly, our data and previously published cases emphasize the need for MOG-IgG antibody testing in patients with a clinical syndrome of non-infectious meningoencephalitis and highlight the importance of recognizing the presentation of MOG-IgG meningoencephalitis as it can often be the initial presentation of relapsing MOGAD.
Recently published diagnostic criteria for MOGAD include CCE as one of the core clinical demyelinating syndromes. 1 However, it is important to recognize that clinical presentation of MOG-IgG meningoencephalitis may vary with unilateral or bilateral involvement as well as meningeal, cortical, and subcortical manifestations.
Limitations of this study include the small cohort size, retrospective design, and dependence upon electronic medical record documentation. Additionally, distinction between ADEM, CCE, and meningoencephalitis cases was not always able to be clarified due to the spectrum of these diagnoses and overlap between them. Its strengths are the detailed assessment of MOG-IgG meningoencephalitis in multiple centers combined with a thorough literature review compiling the current evidence of this disorder. Future studies are needed to better characterize MOG-IgG meningoencephalitis and its clinical course in larger prospective cohorts. Earlier detection of these cases would aid in prompt treatment with corticosteroids which may be associated with improved clinical outcomes.
Conclusions
MOG-IgG meningoencephalitis is a well-recognized manifestation of MOGAD and may present with headaches, seizures, and other inflammatory CNS syndromes including optic neuritis and transverse myelitis. In our cohort and based on literature review, patients tended to be young- to middle-aged adults with slight predilection for males. MRI often revealed unilateral FLAIR hyperintensity, most often with overlying leptomeningeal enhancement, occasionally associated edema and/or diffusion restriction. CSF profiles vary but most frequently show neutrophilic pleocytosis with mild to moderate protein elevation and absent CSF-restricted OCBs. Our findings support the consideration of MOG-IgG testing in the evaluation of non-infectious meningoencephalitis.
Supplemental Material
sj-docx-1-mso-10.1177_20552173261431924 - Supplemental material for Characteristics of myelin oligodendrocyte glycoprotein antibody-associated meningoencephalitis
Supplemental material, sj-docx-1-mso-10.1177_20552173261431924 for Characteristics of myelin oligodendrocyte glycoprotein antibody-associated meningoencephalitis by Aisha Elfasi, Samir Alkabie, Elsa Rodriguez, Mayra Montalvo, Rhaisa Castrodad-Molina, Fernando X Cuascut, George J Hutton, Revere Kinkel, Jennifer Graves and Torge Rempe in Multiple Sclerosis Journal – Experimental, Translational and Clinical
Footnotes
Disclosures
Elfasi has nothing to disclose. Alkabie has nothing to disclose. Rodriguez has served on scientific advisory boards for Sanofi and Amgen. Montalvo served on an advisory board for Horizon and Amgen Therapeutics, she has been a consultant for TG Therapeutics. Castrodad-Molina has no disclosures. Cuascut reports participation in scientific advisory boards for Horizon, Biogen, and Novartis as well as receiving speaker fees from Biogen. Hutton reports participation in scientific advisory boards for Novartis, Genentech, Genzyme, Bristol Myers Squibb, and Autoimmunity Biologic Solutions, Inc and has also received research funding/commercial entities from Genzyme, Biogen, Novartis, Hoffmann La Roche, and MedImmune. Kinkel has received honoraria for non-promotional educational activity from Biogen and served on advisory boards for TG therapeutics and Genentech. Graves has received research support from NMSS, Octave, Biogen, EMD Serono, Novartis, ATARA Biotherapeutics, and ABM. She has served on an advisory board for TG therapeutics and a pediatric clinical trial steering committee for Novartis. She has consulted for Google. Rempe received grant funding from the National Multiple Sclerosis Society. He served on advisory boards for Genentech, Sanofi-Genzyme, EMD Serono, Amgen, TG Therapeutics, and Alexion. He receives contract research support from Genentech, Sanofi-Genzyme, Celgene, and Novartis.
Data availability statement
The data sets generated and analyzed during this study are available from the corresponding author on reasonable request and with necessary approvals.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.