
Abstract
Myelin oligodendrocyte glycoprotein (MOG) antibody–associated disease is a neuroinflammatory disorder (MOGAD) with heterogeneous phenotype including paroxysms of optic neuritis, transverse myelitis, acute disseminated encephalomyelitis, brainstem demyelination, and encephalitis. Fluid-attenuated inversion recovery hyperintense cortical lesions in MOG-associated encephalitis with seizures, or FLAMES, is a manifestation of cerebral cortical encephalitis seen less frequently than other typical MOG antibody–associated disease presentations. Cases of FLAMES are rarer in children, and frequently initially misdiagnosed with infectious meningoencephalitis. Other meningocortical manifestations of MOG antibody–associated disease have been described and likely exist along a continuum. In this retrospective single-center case series, we describe the demographic, clinical, radiographic, laboratory, and electroencephalographic features of 5 children with clinicoradiographic features consistent with the spectrum of MOG-IgG–positive meningocortical syndromes.
Keywords
Myelin oligodendrocyte glycoprotein (MOG) antibody–associated disease (MOGAD) is an inflammatory, demyelinating disorder of the central nervous system, classically associated with episodes of optic neuritis, transverse myelitis, acute disseminated encephalomyelitis, and brainstem demyelination and encephalitis.1,2 Cerebral cortical encephalitis, a rare subset of MOG antibody–associated disease, produces a syndrome of fever, headache, focal seizures, and neurologic deficits, first described in 2017. 3 Cerebral cortical encephalitis can produce a series of magnetic resonance imaging (MRI) findings including T2-weighted fluid-attenuated inversion recovery hyperintense cortical lesions in MOG-associated encephalitis with seizures, known by the moniker FLAMES, as well as a broader spectrum of meningocortical syndromes of cortical and meningeal enhancement.4,5 Cerebral cortical encephalitis is better described in adults, and there is a scarcity of documented pediatric cases. We describe 5 children who presented with MOG-IgG seropositivity and clinicoradiographic features of the meningocortical spectrum.
Patients and Methods
We performed an institutional review board–approved chart review to identify patients with MOG antibody–associated disease and radiographic features consistent with associated meningocortical syndromes. At our institution, serum MOG-IgG1 is tested in children presenting with compatible clinical and radiographic findings of central nervous system inflammatory disease, but not typical for multiple sclerosis. Specific phenotypes of interest are cortical encephalitis and meningoencephalitis of possible noninfectious etiology. Eligible patients had detected MOG-IgG1 with titer ≥1:100 (performed via fluorescence-activated live cell-based assay at Mayo Clinic Laboratories, Rochester, MN), as well as MRI findings demonstrating cortical gray matter or leptomeningeal inflammation in a pattern consistent with MOG antibody–associated disease as determined by a pediatric neurologist (N.V.) and confirmed by a pediatric neuroradiologist (E.G.). We then collected select clinical and laboratory values. Neurophysiological data were collected and confirmed with pediatric epileptologist (S.R.).
Results
Demographics and Clinical Features
Five patients met clinical and radiographic inclusion criteria (Table 1). Median age at onset was 11 years (range 3-12 years) with 3:2 female predominance. Four of 5 patients (80%) were White/non-Hispanic, and 1 patient (20%) was White/Hispanic. Four of 5 patients (80%) presented with prodromal febrile illness, and then progressed to seizures and encephalopathy. Zero patients (0%) developed nonepileptic hyperkinetic movement disorders. One of 5 patients (20%) required intensive care unit admission. Median hospital length of stay was 7 days (range 5-9 days). Infectious workup for patients included cerebrospinal fluid bacterial cultures and viral polymerase chain reaction (PCR) panel, which was negative for all patients.
Summary of Demographics, Clinical, Imaging, Diagnostic, Treatment, and Outcomes Data.
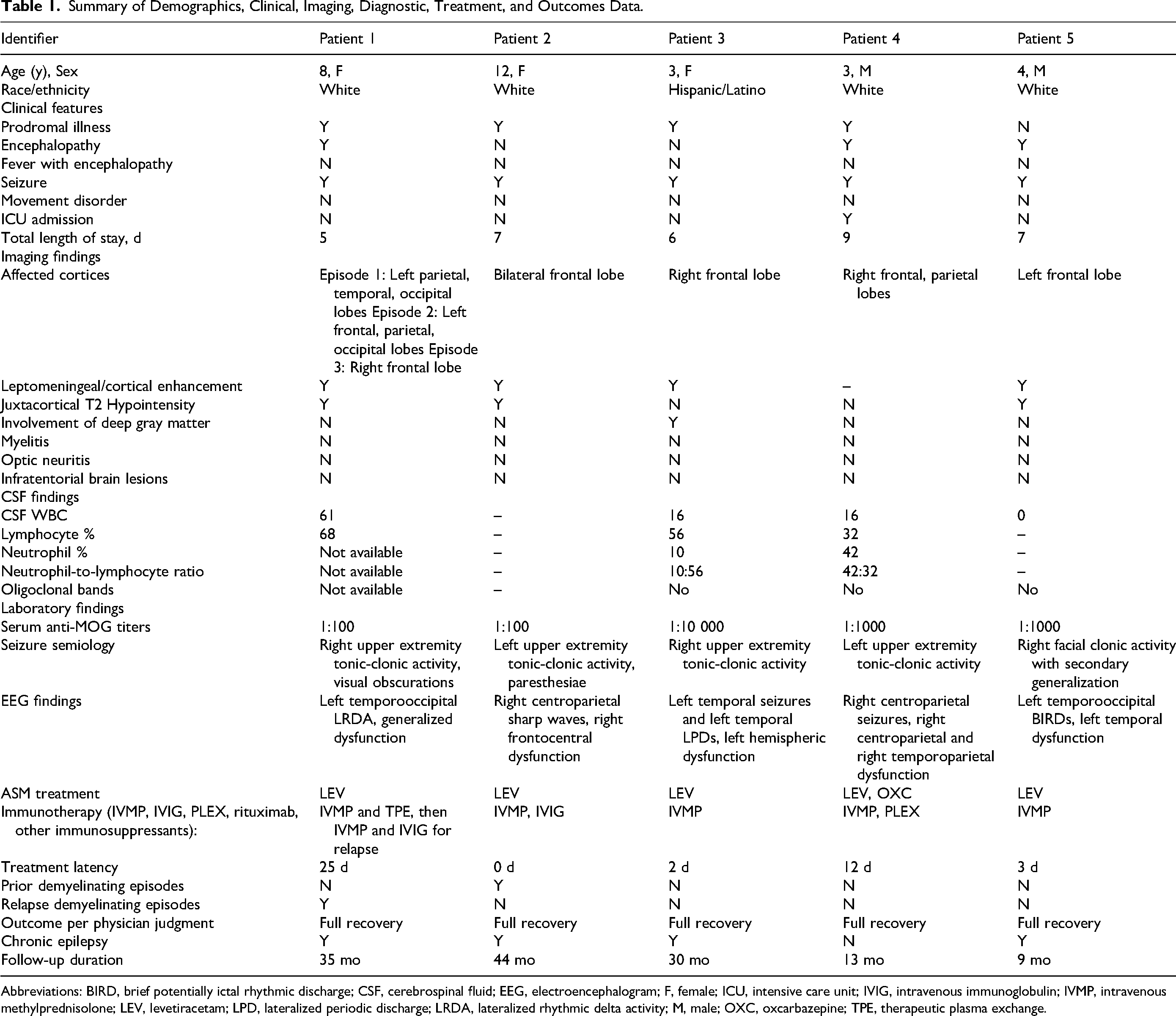
Abbreviations: BIRD, brief potentially ictal rhythmic discharge; CSF, cerebrospinal fluid; EEG, electroencephalogram; F, female; ICU, intensive care unit; IVIG, intravenous immunoglobulin; IVMP, intravenous methylprednisolone; LEV, levetiracetam; LPD, lateralized periodic discharge; LRDA, lateralized rhythmic delta activity; M, male; OXC, oxcarbazepine; TPE, therapeutic plasma exchange.
Radiographic Features
All 5 patients underwent MRI brain with and without gadolinium, with the exception of patient 4, who underwent protocolized imaging without gadolinium with initial clinical concern for stroke (Figure 1). Leptomeningeal enhancement was seen in all patients who received gadolinium and could not be assessed in patient 4. One patient (patient 3) had isolated leptomeningeal enhancement without clear cortical involvement. Patient 2 presented with bilateral involvement, and patient 1 had 3 discrete episodes of cerebral cortical encephalitis involving 3 independent, multifocal cortical regions within both hemispheres. Although 1 patient (patient 3) had deep gray matter involvement, no patients had concurrent optic neuritis, infratentorial brain lesions, or myelitis. Two patients (patients 3 and 5) of 5 (40%) had subcortical involvement; patient 5 had leptomeningeal enhancement and subcortical enhancement, with cortical signal abnormality without associated cortical enhancement. Three of 5 patients (60%) had juxtacortical T2 hypointensities. Arterial spin labeling is not universally performed at our institution, but was performed on patients 1 and 5, with patient 1 demonstrating localized hyperperfusion and patient 5 exhibiting possible hyperperfusion. All 4 cortices were affected but with a frontal predominance. The frontal lobe was affected in 5 of 5 (100%), the parietal lobes in 2 of 5 (40%), the temporal lobe in 1 of 5 (20%), and the occipital lobe in 1 of 5 (20%).
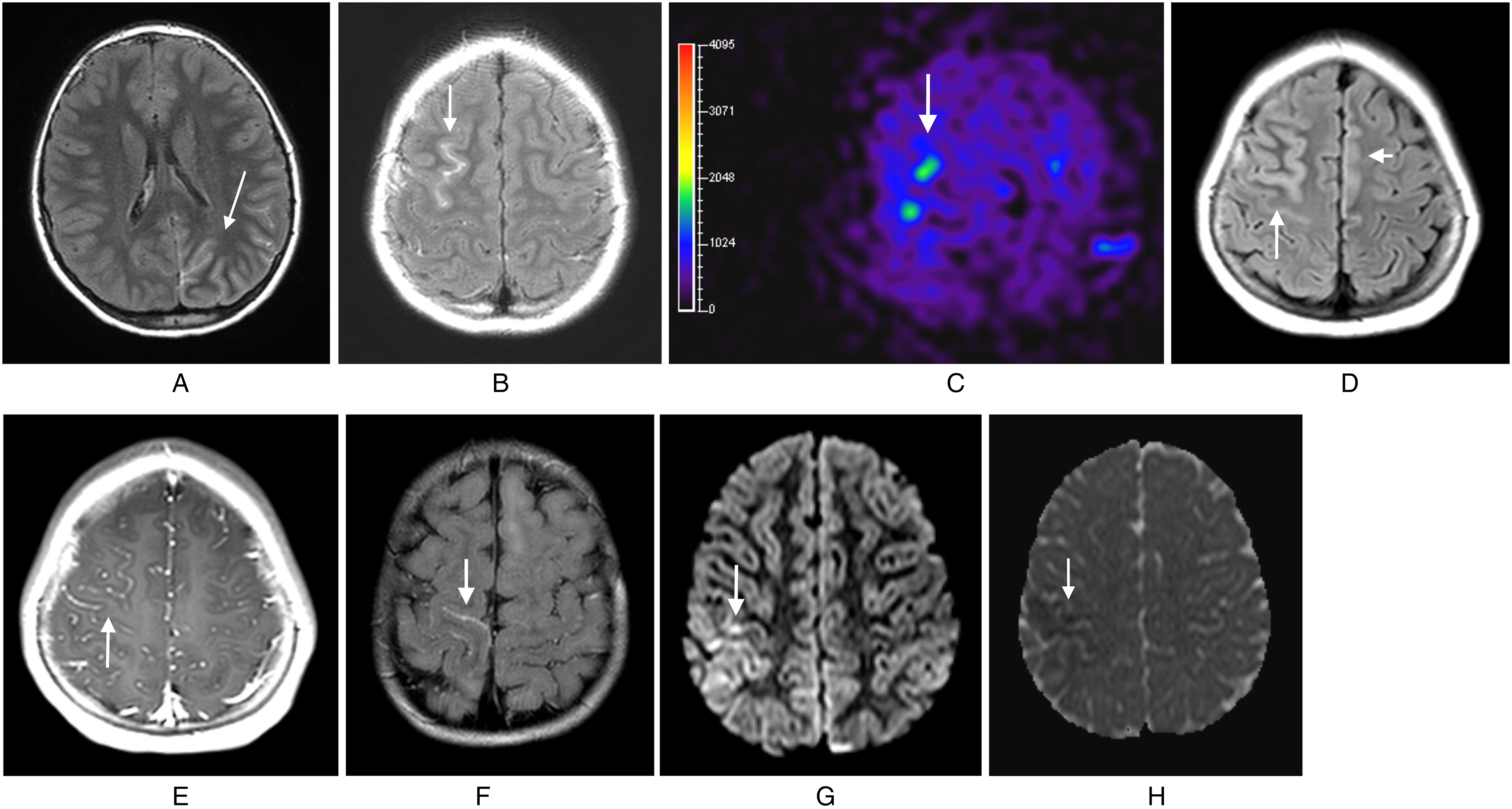
Brain magnetic resonance imaging (MRI) of patients with MOG-IgG–positive meningocortical syndromes. Patient 1: Axial T2-weighted fluid-attenuated inversion recovery (T2-FLAIR) image post-gadolinium (A) during initial presentation demonstrates cortical hyperintensity of the left temporal, parietal, and occipital lobes with associated leptomeningeal enhancement as well as juxtacortical hypointensity. Axial T2-weighted fluid-attenuated inversion recovery image post-gadolinium (B) during relapse demonstrates right frontoparietal leptomeningeal enhancement. Axial arterial spin labeling (ASL) image (C) demonstrates corresponding area of localized hyperperfusion. Patient 2: Axial T2-weighted fluid-attenuated inversion recovery image pre-gadolinium demonstrates bilateral frontal lobe hyperintensity and juxtacortical hypointensity (D), and axial T1 image post-gadolinium demonstrates right frontal leptomeningeal enhancement (E). Patient 3: Axial T2-weighted fluid-attenuated inversion recovery image post-gadolinium (F) demonstrates right superior frontal leptomeningeal enhancement. Patient 4: Axial diffusion-weighted imaging (DWI) image (G) demonstrates right frontoparietal restricted diffusion with corresponding apparent diffusion coefficient (ADC) image (H). Patient 5: Axial T2 pre-gadolinium images (I, J) demonstrate left frontal hyperintensity of the cortex with juxtacortical hyperintensity. Axial T1 post-gadolinium images (K, L) demonstrate left frontal leptomeningeal and subcortical enhancement. Axial ASL image (M) demonstrates possible area of corresponding localized hyperperfusion.
Laboratory Features
Serum MOG titers ranged from 1:100 to 1:10 000. Three of 4 patients (75%) had cerebrospinal fluid pleocytosis, with 2 of 3 (67%) with lymphocytic predominance; the fourth patient did not have cerebrospinal fluid cell count differential. No cerebrospinal fluid–specific oligoclonal bands were found in any of the patients. Patient 2 had undergone lumbar puncture with cerebrospinal fluid studies during initial presentation for optic neuritis, and cerebrospinal fluid studies were not repeated when she represented with cerebral cortical encephalitis.
Neurophysiological Features
Five of 5 patients (100%) presented with clinical seizures, the semiology of which was concordant with cortical regions affected. Seizure semiology included focal motor with preserved awareness in 1 of 4 patients (25%), and focal motor without preserved awareness in 4 of 5 patients (80%). Two of 5 patients (40%) with focal motor seizure without preserved awareness progressed to bilateral involvement. One patient had electroclinical seizures captured on electroencephalogram (EEG), 1 patient had brief potentially ictal rhythmic discharges (BIRDs), and 1 patient had focal interictal sharp waves, but the remainder had no interictal abnormalities. Although all patients had evidence of focal dysfunction, there were no other EEG findings that were specific to these patients. In 5 of 5 patients (100%), EEG and MRI findings showed overlapping affected cortical regions. However, in 2 of 4 patients (50%), the EEG abnormalities were broader than MRI abnormalities, whereas in 2 of 4 patients (50%), the MRI abnormalities were broader than EEG abnormalities.
Treatment and Outcomes
Median treatment latency was 12 days (range 0-25 days). In 3 patients (60%), treatment was delayed because of an initial misdiagnosis of viral meningitis. Five of 5 patients (100%) were treated with corticosteroids. Two of 5 patients (40%) were treated with corticosteroids alone, 1 of 5 patients (20%) treated with corticosteroids and intravenous immunoglobulin, 1 of 5 patients (20%) was treated with corticosteroids and therapeutic plasma exchange, and 1 of 5 patients (20%) was treated with combined corticosteroids, therapeutic plasma exchange, and intravenous immunoglobulin. Levetiracetam was used for acute symptomatic seizures in all 5 patients.
Patient 2 had had prior episode of bilateral optic neuritis, and patient 1 had subsequent bilateral optic neuritis and 2 recurrences of cerebral cortical encephalitis after initial presentation. Patient 1 was subsequently lost to follow-up. Patient 2 is currently being maintained on monthly intravenous immunoglobulin infusions. Patients 3, 4, and 5 have not had recurrence and are not managed on long-term immunomodulation. Four of 5 (80%) have chronic diagnosis of epilepsy requiring long-term antiseizure medication. Four of 4 (100%) with epilepsy are maintained on levetiracetam monotherapy. One patient had previously been on levetiracetam and oxcarbazepine before weaning off antiseizure medications.
Discussion
Detection of antibodies against MOG, a membrane protein of the oligodendrocyte and myelin surface within the central nervous system, via cell-based assay led to recognition of MOG antibody–associated disease as a distinct demyelinating clinical entity, distinct from multiple sclerosis and aquaporin 4 antibody (AQP4-Ab) positive neuromyelitis optica spectrum disorders.2,6 Archetypical manifestations of MOG antibody–associated disease include optic neuritis, transverse myelitis, acute disseminated encephalomyelitis, and brainstem demyelination and encephalitis. Although both adults and children with MOG antibody–associated disease often present with optic neuritis, younger children in particular are more likely to present with acute disseminated encephalomyelitis, but they are more likely to have better recovery with lower rates of relapse. 1 The clinical presentations of MOG antibody–associated disease in children are much more heterogenous compared to adults, with phenotypic presentations influenced by age. Acute disseminated encephalomyelitis is the most common phenotype as the initial presentation of MOG antibody–associated disease in pediatric patients overall, but further stratification by age suggests that acute disseminated encephalomyelitis is more common in children less than 10, whereas optic neuritis is more common for those aged 10 years and older.1,7
Ogawa et al 3 initially described 4 patients with MOG antibody–positive episodes of unilateral cerebral cortical encephalitis with seizures associated with T2-weighted fluid-attenuated inversion recovery hyperintensities on MRI, which was later given the moniker FLAMES. 8 This syndrome has since been better described, and cerebral cortical encephalitis is better understood to be a clinicoradiographic spectrum, classically associated with preceding fever, headache, focal seizures, and neurologic deficits, with associated MRI abnormalities including cortical T2-weighted fluid-attenuated inversion recovery hyperintensities.8,9 Although initially described as unilateral, cases of bilateral cortical involvement and meningeal inflammation have been described with variable terminology, including bilateral medial frontal cerebral cortical encephalitis (BFCCE), 10 bilateral parafalcine cortical and leptomeningeal impairment (BPCLI), 11 fluid-attenuated inversion recovery–variable unilateral enhancement of the leptomeninges (FUEL), 5 and MOG antibody–associated aseptic meningitis (MOGAM). 12 Recent consensus statement regarding MOG antibody–associated disease specifically included cerebral cortical encephalitis, including both unilateral and bilateral cortical T2-weighted fluid-attenuated inversion recovery hyperintensities, as well as cortical and leptomeningeal enhancement, even in the absence of T2-weighted fluid-attenuated inversion recovery changes, within the same spectrum. 2
Review of prior cases suggests that cerebral cortical encephalitis presents with seizures, headache, and encephalopathy in most patients, and fever and focal neurologic deficits in a substantive minority,13,14 which is consistent with our patients. Given shared presenting symptomatology, initial confusion for infectious meningoencephalitis is not uncommon, 14 as occurred in 3 of our 5 patients. Patients frequently, but not always, have a prior history of a central nervous system demyelinating syndrome, such as optic neuritis or acute disseminated encephalomyelitis. 8 One of our patients did have a history of bilateral optic neuritis, but for the others, this was the first clinical manifestation of MOG antibody–associated disease.
Cerebrospinal fluid studies typically show lymphocytic pleocytosis and absence of cerebrospinal fluid-specific oligoclonal bands. 13 A majority of our patients had cerebrospinal fluid pleocytosis, and a majority of those had lymphocytic predominance. No patients had cerebrospinal fluid–specific oligoclonal bands. Although serum MOG-IgG positivity is universal in cerebral cortical encephalitis, the full significance of its titer is not fully elucidated. The positive predictive value (PPV) appears to be titer dependent and is even higher in children. 15 Without statistical significance, there is a suggestion that higher titer value results in longer duration of headache and fever, a higher frequency of seizures, and a greater degree of visual impairment.8,13
Although cortical T2-weighted fluid-attenuated inversion recovery hyperintensities were initially described, cortical and leptomeningeal gadolinium enhancement has also been demonstrated in a sizable number as well. 4 All 4 of our patients who underwent MRI with gadolinium demonstrated leptomeningeal contrast enhancement, suggesting this is a prominent feature in children. Cerebral cortical encephalitis was initially described as unilateral cerebral cortical encephalitis 3 as the majority of patients presented with unilateral involvement, but recent guidelines acknowledge that cerebral cortical encephalitis can be both unilateral and bilateral. 2 The majority (80%) of our patients presented with unilateral involvement, but 1 patient did present with bilateral involvement, and a second patient presented with 3 discrete episodes of unilateral involvement affecting 3 discrete regions in both hemispheres. One patient exhibited extension of cortical edema into the surrounding white matter, as has been described in prior reports. 16 These diverse radiographic findings in the setting of unifying clinical presentation and MOG-IgG seropositivity help support the notion of a broader meningocortical spectrum associated with MOG antibody–associated disease in children, in keeping with recent consensus guidelines. 2
In additional to cortical and leptomeningeal abnormalities, adjacent subcortical T2-weighted fluid-attenuated inversion recovery hypointensities were seen in a majority of our patients. Previous studies have also demonstrated this and suggested that cortical dysfunction impeding glymphatic flow may produce a transient decrease in interstitial fluid.9,14 Localized hyperperfusion of cortical lesions is thought emblematic of cerebral cortical encephalitis, such as seen with single proton emission computed tomography.3,13 Arterial spin labeling has been previously shown localized hyperperfusion associated with cerebral cortical encephalitis, 16 and our cohort suggests this may be a useful supplement. Future studies are required to better characterize the relation between hyperperfusion seen on arterial spin labeling and meningocortical syndromes.
Prior reports suggested that frontal, parietal lobes are involved more frequently than the temporal and occipital lobes, and typically seizure semiology or focal neurologic deficits correspond to affected cortical areas.4,13 In our cohort, there was a frontal predominance: all 5 patients had involvement of the frontal lobes, whereas 2 also had parietal lobe involvement, 1 had temporal lobe involvement, and 1 had occipital lobe involvement.
Although classically cerebral cortical encephalitis has been described as relatively benign and generally steroid-responsive,8,13 a more severe phenotype of children has been described.8,14,16 This focal cerebral cortical encephalitis is defined by the presence of bilateral, multifocal, asymmetric cortical gray matter diffusion restriction with absence of features consistent with white matter–predominant syndromes, and absence of vascular distribution of diffusion restriction. 16 None of our patients met radiographic criteria for focal cerebral cortical encephalitis.
Focal seizures were ubiquitous in our cohort, consistent with prior studies, with a subset evolving with bilateral involvement and progressing to status epilepticus. 17 All of our patients had focal dysfunction on EEG, in contrast to prior studies suggesting that only a minority have focal dysfunction. 17 Focal dysfunction in our patients corresponded to MRI abnormalities and presumed seizure nidus. In 2 patients, there was a broader field of electrographic involvement than seen on MRI, and in 2 other patients, there was a broader field of radiographic involvement than seen on EEG.
Treatment of acute symptomatic seizures with antiseizure medications in the setting of cerebral cortical encephalitis is ubiquitous, although prior studies have suggested that antiseizure medication use does not significantly reduce the incidence of seizures, whereas immunotherapy was associated with decreased risk of recurrent seizure. 18 Optimal duration of antiseizure medication use in these patients is still unclear. Montalvo et al noted that all patients had a ≥50% reduction in seizure frequency with antiseizure medication therapy and immunotherapy, and 91% were seizure-free at their last follow-up. 17 They did note that 3 patients developed drug-resistant epilepsy, and it is worth noting that in a majority of our patients, there was a chronic need for maintenance antiseizure medications. However, all of our patients are seizure-free on antiseizure medication monotherapy, and none have drug-resistant epilepsy. The long-term risk of epilepsy in children with cerebral cortical encephalitis may be more heterogenous than previously thought.
Conclusion
The clinical and radiographic presentations of our patients are similar to those previously described, suggesting that children with MOG antibody–associated disease meningocortical syndromes present along a similar phenotypic spectrum as adults, but with some caveats. Leptomeningeal enhancement seems an especially prominent feature. Our cohort also demonstrates that bilateral involvement and relapse, although less common, do also occur in children. Optimal duration of antiseizure medication therapy in concert with immunotherapy needs to be thoroughly investigated.
Footnotes
Author Contributions
RBC and NHV contributed to conception and design; RBC, KB, EBG, SBR, and NHV contributed to acquisition, analysis, and interpretation of data, drafted a significant portion of the manuscript; RBC, EBG, SBR, and NHV critically revised the manuscript; All authors gave final approval for the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
The Vanderbilt University Medical Center institutional review board waived the need for ethics approval and the need to obtain consent for the collection, analysis and publication of the retrospectively obtained and anonymized data for this noninterventional study.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.