
Abstract
Background
Individuals with neuromyelitis optica spectrum disorder (NMOSD) often suffer from severe, disabling, and treatment-refractory neuropathic pain. Transcutaneous electrical nerve stimulation (TENS) therapy is a non-invasive, pain-modifying device.
Objective
To determine whether TENS therapy is safe, tolerable, and effective for neuropathic pain in patients with NMOSD.
Methods
We conducted a four-week, randomized, double-blind, sham-controlled, remote trial of TENS in patients with NMOSD who have neuropathic pain, followed by a 12-week open-label extension period. The difference in the Numeric Rating Scale current pain scores between 0 weeks and 4 weeks was the primary outcome measure.
Results
Forty-six patients (23 per arm) were enrolled in this trial, of which 40 were included in the primary analysis (four in the intervention arm and two in the sham arm withdrew prior to assessment of the primary outcome). Both the sham and intervention arms demonstrated significant decreases in average pain, worst pain, and current pain rating between baseline and 4 weeks, but there was no significant difference between the two arms.
Conclusions
In conclusion, there was no demonstrated benefit of TENS over sham TENS treatment, however, both arms demonstrated significant decreases in reported pain between baseline and 4 weeks. This trial is registered with ClinicalTrials.gov, NCT04614454.
Keywords
Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is a chronic relapsing autoimmune disease of the central nervous system (CNS) that preferentially targets the optic nerves and spinal cord. 1 Pain is a disabling component of the disease with up to 86% of patients reporting central neuropathic pain (CNP) characterized by burning, shooting, or tingling sensations. 2 NMOSD lesions in the spinal cord are characteristically long and destructive, and pain is more prevalent in NMOSD than in most other neurological diseases. 3 Research on the impact of persistent pain on quality of life in NMOSD has found that patients with CNP experience more depression, less enjoyment of life, and more difficulty with ambulation than the general population. 4 Currently, there is no standard of care for CNP treatment and off-label medications typically used for multiple sclerosis or peripheral neuropathy are often insufficient to control NMOSD-related pain. 5 In fact, research shows that increased dosages of commonly prescribed pain medications may lead to worsened cognitive difficulties and fatigue rather than reduced pain. 2
Transcutaneous electrical nerve stimulation (TENS) therapy is a non-invasive pain-modifying intervention that utilizes regularly changing electric pulses to stimulate nerves so that the experience of pain is blocked via transcutaneous electrical stimulation of ascending sensory fibers with the goal of re-organizing maladaptive signaling pathways. 6 This neuromodulatory therapy has been investigated for the treatment of persistent peripheral neuropathic pain in several conditions including chemotherapy-induced neuropathy, 7 post-herpetic neuralgia, 8 and post-surgical neuropathic pain. 9
For patients with NMOSD and other conditions that cause pain originating in the CNS, non-pharmacological devices would be an appealing alternative or addition to pain medications. A previous trial in this population with a large-scale transcutaneous electronic nerve stimulator, the Scrambler® TENS device, showed positive results. 10 However, the Scrambler required office-based operation by a professional technician, thereby limiting its usefulness for NMOSD patients who often have a disability and chronic pain that limits mobility. The goal of this trial was to investigate a portable, wearable, non-invasive TENS unit that could be safely employed at home for the treatment of neuropathic pain in NMOSD.
Methods
Trial design
This study was a prospective, double-blind, randomized, sham-controlled clinical trial. We enrolled 46 participants with NMOSD who experienced regular pain and randomized each participant at a 1:1 allocation ratio to the following two treatment groups: (1) active TENS device; and (2) sham TENS device. The primary experimental, parallel arm period of the trial occurred during the first 4 weeks, with the primary outcome assessed at 4 weeks. This was followed by a 12-week open-label extension. We adhered to CONSORT reporting guidelines. 11
Participants
Participants were recruited via social media outreach and directly in the clinic at the Neuroimmunology Clinic and Research Laboratory at Massachusetts General Hospital. The trial was entirely virtual, and the research coordinator screened potential participants by phone. Inclusion criteria included age 18+ years, diagnosis of NMOSD, presence of aquaporin-4 immunoglobulin (AQP4 IgG) serum antibodies, pain lasting ≥ 3 months attributable to a previous inflammatory spinal cord lesion, and pain rated at a level of ≥ 4 on an 11-point Numeric Rating Scale (NRS). 12 Exclusion criteria included spinal cord relapses or surgery to treat a pain-related condition within 6 months of enrollment, presence of neuropathic pain due to non-NMOSD-related identified causes, and presence of metal implants or spinal cord stimulators.
Interventions
Written informed consent was obtained from each participant. After determination of eligibility, participants were randomized to receive either an active TENS unit or a sham TENS unit. The self-administered, take-home Quell FLEX unit is a TENS device based on a Quell 2.0 device modified for study use. It utilized biphasic stimulation pulses (first phase 100 μs and second phase 180 μs), stimulation frequency modulated between 2 and 125 Hz, and a target maximum intensity (mA) maintained from 2 to 10 Hz and then scaled down to 70% of maximum as frequency increase from 10 to 125 Hz (for detailed specifications, see https://www.quellrelief.com/the-quell-system/tech-specs-2-0/). Participants controlled the unit through a mobile application on their phones that was Bluetooth-connected to the device. Usage and intensity data were collected remotely by the product manufacturer and shared with the study team. The TENS unit itself was connected by cable to a butterfly shaped gel pad which the participant placed over the site on the spine, with the level of placement informed by the level of the spinal cord lesion and by the location of maximal pain. The target maximum intensity, or the maximal stimulation intensity level that was tolerated without being uncomfortable, was calibrated for each participant during the first use of the unit.
The device manufacturer was responsible for randomizing the sham and active devices using blocks of four and sending the appropriate device directly to each participant. Both researchers and participants were blinded to the randomization scheme until after the completion of the study. The lights, charging equipment, and app connectivity for the sham TENS were the same as for the active TENS, however, the sham device administered only 2 min of electrical stimulation three times during an hour-long session (at 0, 30, and 58 min), as opposed to administering the stimulation during the full hour session for the active device. All participants were told to wear the device for at least 5 hours daily, which translated to 3 hours of active therapy (or sham), as the TENS device is programmed to turn itself on and off every other hour while in use. Participants received a new active TENS device at the end of the 4-week experimental phase and the beginning of the open-label phase.
Outcomes
The primary objective was to determine the difference in current pain on the NRS score between the baseline screening call and at week 4 in the treatment group compared with the same difference in the placebo or sham group. Key predefined secondary outcome measures included the difference between groups in change in average pain and worst pain on the NRS between the baseline screening call and at week 4, the difference in hours of device usage, the difference in change from baseline to week 4 quality of life as measured by the short form 36 health survey (SF-36), 13 and the difference in treatment-related adverse events.
Sample size
Power analyses determined that a sample size of 19 subjects per arm gave the study a > 90% power to detect a difference between groups of 2.0 points on the 11-point NRS, when assuming a standard deviation (SD) of 2.0. To account for a 20% dropout rate, four subjects were added to each arm for a total of 23 subjects, or 46 in total.
Statistical methods
Differences between groups at baseline were assessed, and univariate descriptive analyses were performed on all the dependent variables. Perceived improvement in pain at 4 weeks was assessed using a two-sample t-test of NRS pain scores between the active and sham treatment groups using a type I error rate of 0.05 (two-sided). The primary and secondary analyses were conducted using a modified intention-to-treat approach, with all participants who completed assessments at baseline and week 4 included in the analysis, regardless of their device usage. Statistical analysis was performed using Stata/SE, version 17.0.
Registration
This study was approved by the Mass General Brigham Institutional Review Board and is registered on ClinicalTrials.gov (#NCT04614454).
Results
Participant flow
Fifty-nine individuals responded to online or in-person information about the study and were virtually screened for trial participation (Figure 1). Forty-six individuals with pain and AQP4-IgG positive NMOSD were enrolled. Of the 13 individuals who were screened but not ultimately enrolled, three did not reply to follow-up contact, and 10 did not meet eligibility criteria. All individuals who completed their baseline and week 4 assessment were included in the analysis as randomized, regardless of actual device usage or further participation in the trial. Twenty-one participants in the sham arm and 19 in the TENS arm met these criteria. Of the six individuals who withdrew prior to week 4, two were in the sham arm (one withdrew due to a treatment-related adverse event (TRAE) and one for unrelated reasons) and four individuals were in the treatment arm (two withdrew due to TRAEs and two for unrelated reasons). Participants who withdrew reported lower initial pain burdens, as indicated by their baseline SF-36 pain subscale scores, compared to those who completed the study (45.4 vs. 25.5; p = 0.029, with higher scores reflecting lower reported pain burdens), however, no other significant differences were observed between the groups (see Supplemental Table 1). Recruitment was conducted from January 2021 to April 2022, when enrollment was complete.
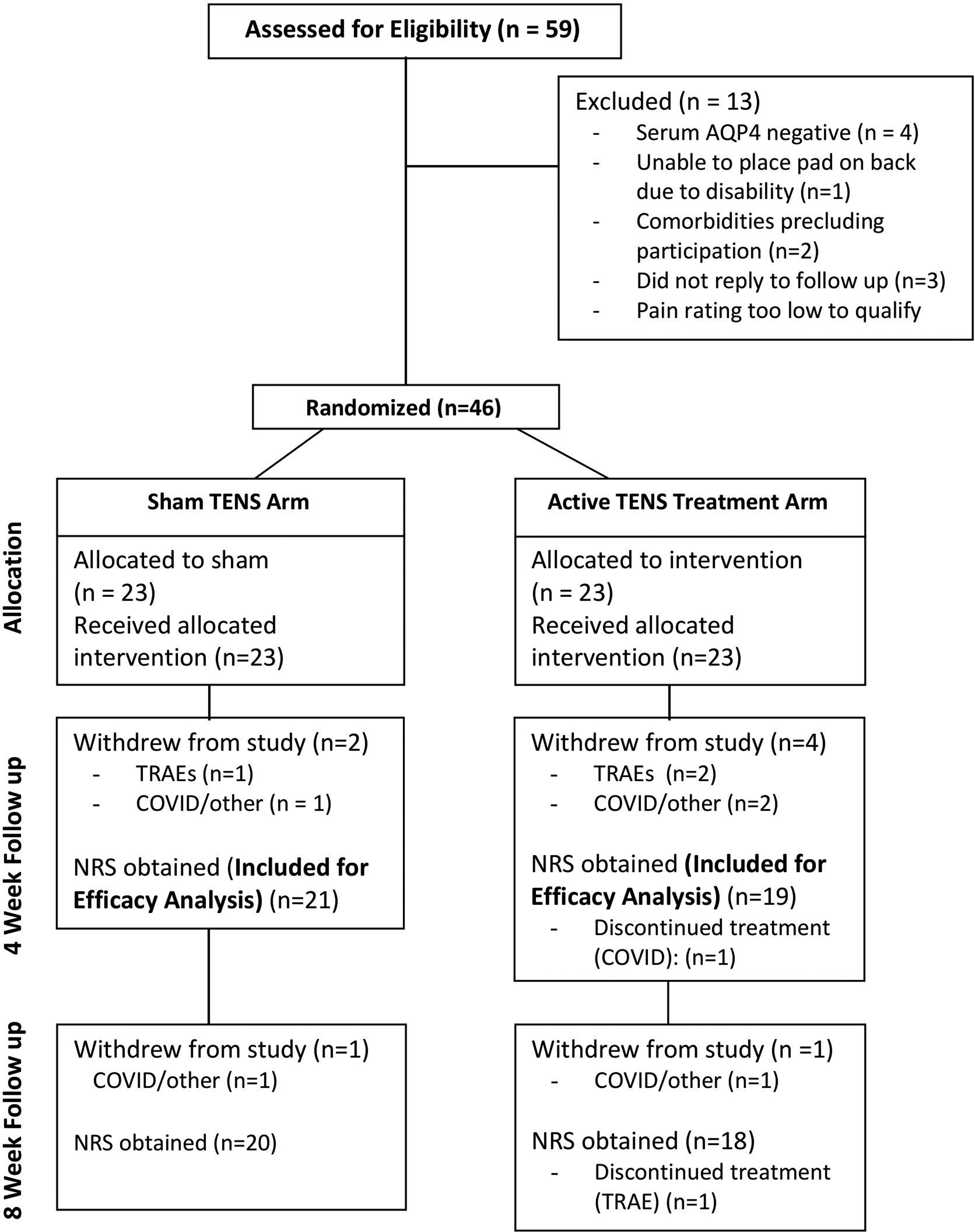
Consolidated standards of reporting trials (CONSORT) participant flow diagram.
Baseline data
Forty females and six males participated in the trial, with an even distribution of sex across treatment groups. Thirty participants were white, eight were black and eight were Hispanic. The average participant age was 52.3 years (SD = 14.2). Thirty three of 46 (71.7%) participants were taking at least one concomitant prescription pain medication, 29 (63%) reported that their ambulation was restricted due to pain, and 24 (52.2%) had previously used some version of TENS therapy. The baseline current pain rating in the active TENS arm was higher than in the sham arm (6.7 vs. 5.5, p = 0.053), although this difference did not reach statistical significance. Other baseline characteristics were similar across treatment arms (Table 1).
Baseline characteristics by treatment group.

TENS: transcutaneous electrical nerve stimulation; SF-36: 36-item short form survey. Data are presented as mean (SD) for continuous measures, and n (%) for categorical measures.
Effectiveness outcomes
1. Quantitative pain measures: The primary outcome measure, change from baseline in reported current pain at week 4, was greater in the active TENS arm compared to the sham arm, but this difference was not significant (2.16 ± 2.24 NRS points vs. 1.29 ± 2.34 NRS points, p = 0.2377) (Table 2). There were no significant between-group differences detected in any NRS pain scores at week 4, regardless of whether the change from baseline (Table 2) or absolute score values (Figure 2) were used.
Both study arms, however, demonstrated a significant decrease in current pain, average pain, and worst pain scores between the baseline and 4-week assessment, and between baseline and the 8-week assessment, with the exception of the worst pain scores in the active arm between baseline and week 4 (p = 0.058) (Table 3).
2. Quality of life measures. Participants in the study had notably lower (i.e. worse) baseline scores than the general population in the subscales relating to physical function, role limitations due to physical health, energy/fatigue, pain, and general health perception (Table 1, Supplemental Table 2). Participants in the active TENS arm reported a quantitatively greater improvement in quality of life from baseline to week 4 on the aggregate summary score and on all eight subscale scores of the SF-36 compared to the sham arm, however, these differences were not significant (Table 2). As with the quantitative NRS scores, both study arms demonstrated a significant improvement across the SF-36 summary score from week 0 to week 4 and from week 0 to week 8 (Table 3). Study participant comments, systematically recorded and aggregated in Supplemental Table 4, mirrored the results seen with the quantitative pain and quality of life measures. Specifically, many participants reported improvement or changes in their pain, regardless of their study arm or the study period, although there were more positive comments during the open-label extension period and in the active arm. Themes discussed by participants included improvements in walking, improvements in strength and other functions, improvements in sleep, helpfulness of the device only while “on,” “relaxing” effects, decreases in required pain medications, usefulness for travel, as well as lack of change in pain or even worsening of pain in some cases.

NRS-P scores by treatment group. Actual Numeric Rating Scale (NRS) mean scores for current pain, average pain, and worst pain over both the parallel arm experimental period (weeks 0–4) as well as the initial open-label extension weeks (weeks 4–8).
Differences between baseline and 4-week NRS and SF-36 by treatment group.

TENS: transcutaneous electrical nerve stimulation; NRS: Numeric Rating Scale; SF-36: 36-item short form survey. Data are presented as mean difference ± SD, with a two-sample t-test conducted to compare between-group differences. Positive scores indicate improvements (i.e. lower score for NRS and higher score for SF-36), and negative scores indicate worsening.
NRS-P and SF-36 at baseline, 4-week, and 8-week follow-up by treatment group.

NRS: Numeric Rating Scale; TENS: transcutaneous electrical nerve stimulation; SF-36: 36-item short form survey. Data are presented as mean NRS score at time point ± SD, with a two-sample t-test conducted to compare between-group differences. Scores are compared between baseline and week 4 (first p-value), and then between baseline and week 8 (second p-value) in each treatment group.
Denotes p < .05.
SF-36 summary score results reported, with higher scores indicating higher quality of life.
Acceptability and safety measures
Participants in the active TENS arm reported 16 TRAEs compared to 14 TRAEs in the sham arm. There were no treatment-related serious adverse events in either study arm. Participants in both arms voiced design-related device problems including problems with the pads or wires coming off, difficulty placing the pad, and inconvenience of wearing the device, with 15 individual complaints in the active arm, and 31 individual complaints in the sham arm (Table 4).
Treatment-related adverse events by treatment group.

TENS: transcutaneous electrical nerve stimulation.
Indicate adverse events attributed to the device and leading to device use discontinuation by participants, but not thought to be related by investigators.
Average daily hours of device usage (measured remotely via the device) through week 4 did not differ between sham and active TENS arms (2.86 ± 1.4 h vs. 2.87 ± 1.4 h, p = 0.9713). The hypothesis that nonadherence rates would differ between the sham and active TENS arms of the study was not borne out, with roughly identical usage seen in both arms throughout the study (Supplemental Table 3).
Sensitivity analyses
Post-hoc sensitivity analyses were conducted to evaluate whether the study conclusions were robust to a per-protocol analysis incorporating actual device usage data. We used linear regression to evaluate the impact of study arm assignment on pain scores while controlling for hours of device usage and baseline pain scores, and study conclusions remained the same. There were also no significant differences in results between those who had previously used TENS therapy and those who had not. For additional sensitivity analysis, we used mixed effects with repeated measures model to analyze the primary NRS outcome and secondary NRS outcomes while incorporating all NRS scores from week 0 to 4 rather than just week 4 NRS scores, and this also did not change the study conclusions.
Discussion
This double-blind, randomized, sham-controlled clinical trial in individuals with NMOSD and neuropathic pain demonstrated that the use of both the active stimulation TENS unit as well as the minimal stimulation sham TENS units significantly decreased patient-reported pain and improved health-related quality of life measures. Improvements in quality of life measured by the SF-36 were quantitatively greater in the active arm compared to the sham across all domains as well as the aggregate score, however, none of these differences were significant. Anecdotally, many participants reported the efficacy of the device only when it was actively stimulating, or when used on an “as-needed” basis, such as for travel, or at times of more significant pain, however, this was not systematically captured in our quantitative study outcomes (Supplemental Table 4). No serious treatment-related adverse reactions occurred in either arm.
There are several potential explanations for the benefit perceived in both arms of the study. First, a significant placebo effect in central neuropathic pain is well-documented, has tended to increase in pain trials over time, and likely played a role.14,15 Furthermore, the effect of a sham intervention as used in this study may be somewhat greater than a pharmacologic placebo. 16 Alternatively, it is possible that the minimal stimulation from the sham unit had a physiologic effect and that our study was in fact testing a higher-intensity intervention and a lower-intensity intervention rather than an active and inactive intervention. Given the dysesthesia and hyperalgesia that can occur with transverse myelitis, it is possible that even the sensation of the patch on the skin and the infrequent stimulation had an impact on pain. A prior study of the same unit for the treatment of fibromyalgia-related pain also showed a significant improvement in perceived pain in both study arms, but no difference between active and sham TENS. However, there was notably a between-group difference detected specifically for the subgroup that had high pain sensitivity on quantitative sensory testing. 17 Our study did not evaluate pain sensitivity, so we may have missed this signal. Our study's results differed from those of a prior study using Scrambler therapy which found evidence of efficacy for the treatment of neuropathic pain in this population; this discrepancy may be attributed to the more intensive nature of in-person Scrambler therapy sessions administered in precise locations daily by a technician, as compared to the remote, self-administered TENS unit used in this study. 10
This study had several limitations. First, the study protocol did not explicitly ask participants to rate their pain while wearing the device, although, in their subjective comments, participants frequently reported pain relief only during the time that their device was active (Supplemental Table 4—“Helpful when ‘On’” theme), or that they found the device helpful for specific situations and environments. Second, although participants were encouraged to maintain a constant dosage of their other pain medications throughout the study, this was not systematically assessed, and many participants reported decreasing their pain medication dosages or frequency of as-needed pain medication as a benefit of the TENS device (Supplemental Table 4—“Decreasing pain medication” theme). These decreases may have occurred asymmetrically between study arms, resulting in an apparent lack of difference between arms. Additionally, many participants reported multifactorial pain, and had difficulty distinguishing their neuropathic pain from muscle stiffness-related pain or other types of pain that may have been less affected by the TENS unit. We also do not have data on whether participants who found the therapy (either sham or active) beneficial continued to have sustained relief in their pain after study intervention discontinuation, although this would have important implications for treatment recommendations. Finally, the study was conducted during the pandemic, and a significant proportion of subjects were infected with SARS-CoV-2 which impacted their pain levels and adherence to the study, likely resulting in lower overall device usage rates and greater fluctuations in pain levels.
In summary, while this clinical trial found no significant difference in pain ratings between active and sham TENS treatment arms in individuals with NMOSD-related pain, participants in both study arms reported significant improvements in pain after 4 weeks of device use. Wearable, portable TENS devices may be considered as an adjunctive pain relief tool for individuals experiencing CNP related to NMOSD who do not tolerate or have inadequate pain relief with pharmacologic options. Future studies utilizing TENS devices for pain relief should investigate the role of different stimulation regimens (sham, higher, and lower intensity) on efficacy, consider the incorporation of quantitative sensory testing, measure long-term pain levels after treatment discontinuation, and also include measures to assess pain relief with the devices specifically while they are actively stimulating.
Supplemental Material
sj-docx-1-mso-10.1177_20552173241301018 - Supplemental material for A transcutaneous electrical nerve stimulation device for the relief of neuropathic pain in NMOSD: A randomized, double-blind, sham-controlled trial
Supplemental material, sj-docx-1-mso-10.1177_20552173241301018 for A transcutaneous electrical nerve stimulation device for the relief of neuropathic pain in NMOSD: A randomized, double-blind, sham-controlled trial by Anastasia Vishnevetsky, Gabriela Romanow and Michael Levy in Multiple Sclerosis Journal – Experimental, Translational and Clinical
Supplemental Material
sj-docx-2-mso-10.1177_20552173241301018 - Supplemental material for A transcutaneous electrical nerve stimulation device for the relief of neuropathic pain in NMOSD: A randomized, double-blind, sham-controlled trial
Supplemental material, sj-docx-2-mso-10.1177_20552173241301018 for A transcutaneous electrical nerve stimulation device for the relief of neuropathic pain in NMOSD: A randomized, double-blind, sham-controlled trial by Anastasia Vishnevetsky, Gabriela Romanow and Michael Levy in Multiple Sclerosis Journal – Experimental, Translational and Clinical
Supplemental Material
sj-docx-3-mso-10.1177_20552173241301018 - Supplemental material for A transcutaneous electrical nerve stimulation device for the relief of neuropathic pain in NMOSD: A randomized, double-blind, sham-controlled trial
Supplemental material, sj-docx-3-mso-10.1177_20552173241301018 for A transcutaneous electrical nerve stimulation device for the relief of neuropathic pain in NMOSD: A randomized, double-blind, sham-controlled trial by Anastasia Vishnevetsky, Gabriela Romanow and Michael Levy in Multiple Sclerosis Journal – Experimental, Translational and Clinical
Supplemental Material
sj-docx-4-mso-10.1177_20552173241301018 - Supplemental material for A transcutaneous electrical nerve stimulation device for the relief of neuropathic pain in NMOSD: A randomized, double-blind, sham-controlled trial
Supplemental material, sj-docx-4-mso-10.1177_20552173241301018 for A transcutaneous electrical nerve stimulation device for the relief of neuropathic pain in NMOSD: A randomized, double-blind, sham-controlled trial by Anastasia Vishnevetsky, Gabriela Romanow and Michael Levy in Multiple Sclerosis Journal – Experimental, Translational and Clinical
Supplemental Material
sj-docx-5-mso-10.1177_20552173241301018 - Supplemental material for A transcutaneous electrical nerve stimulation device for the relief of neuropathic pain in NMOSD: A randomized, double-blind, sham-controlled trial
Supplemental material, sj-docx-5-mso-10.1177_20552173241301018 for A transcutaneous electrical nerve stimulation device for the relief of neuropathic pain in NMOSD: A randomized, double-blind, sham-controlled trial by Anastasia Vishnevetsky, Gabriela Romanow and Michael Levy in Multiple Sclerosis Journal – Experimental, Translational and Clinical
Supplemental Material
sj-docx-6-mso-10.1177_20552173241301018 - Supplemental material for A transcutaneous electrical nerve stimulation device for the relief of neuropathic pain in NMOSD: A randomized, double-blind, sham-controlled trial
Supplemental material, sj-docx-6-mso-10.1177_20552173241301018 for A transcutaneous electrical nerve stimulation device for the relief of neuropathic pain in NMOSD: A randomized, double-blind, sham-controlled trial by Anastasia Vishnevetsky, Gabriela Romanow and Michael Levy in Multiple Sclerosis Journal – Experimental, Translational and Clinical
Footnotes
Acknowledgements
The authors would like to thank Dr Robert Jamison for sharing his experience with studying the Quell TENS unit with the study team. The authors would additionally like to thank all the patients who participated in the clinical trial.
Data availability statement
The complete datasets generated and analyzed in this clinical trial are available from the corresponding author upon reasonable request.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The manufacturer, Neurometrix, provided the Quell TENS units free of charge. Funding for the study conducted was provided by the Harvard Medical School Ruth and Maurice Freeman Award for Pain Neurology Research. AV was supported in part by a grant from the National Multiple Sclerosis Society while conducting analysis for this study. She was additionally funded in part with Federal funds from the National Institute of Neurological Disorders and Stroke, National Institutes of Health, as a NeuroNext fellow. This work was conducted with support from Harvard Catalyst | The Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences, National Institutes of Health Award UL1 TR002541) and financial contributions from Harvard University and its affiliated academic healthcare centers. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University, and its affiliated academic healthcare centers, or the National Institutes of Health.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.