
Abstract
Background
Ocrelizumab and rituximab are frequently used treatments for multiple sclerosis (MS). Data on switching from rituximab to ocrelizumab is limited.
Objectives
To assess the frequency, severity, and factors of infusion related reactions (IRRs) in patients with MS who switch from rituximab to ocrelizumab, compared to those who stay on rituximab.
Methods
Prospective study on MS patients aged 18–65, on rituximab for at least 2 cycles, who either switched to ocrelizumab (switch group) or stayed on rituximab (comparator group) (n = 100 each). Participants were followed for IRRs, safety, and tolerability over 12 months.
Results
The proportion of IRRs in patients who continue on rituximab (14%) were similar to those who switched to ocrelizumab on Day 1 (14%; p = 1.000) and Week 24 (12%; p = 0.647) but higher than at Day 15 (4%; 0.005). The risk of IRRs for the switch group was associated with the presence of B cells (CD19 and/or CD20 counts ≥1%) increasing by 5.01 (1.49, 16.82) times on Day 1 (p = 0.007). Antidrug antibodies to ocrelizumab were not associated with IRRs. No other safety concerns were identified in switching to ocrelizumab.
Conclusion
IRRs are similar between both groups, which suggests that it is safe to switch from rituximab to ocrelizumab.
Introduction
The treatment of multiple sclerosis (MS) with B-cell directed therapies that target CD20 have proven beneficial. 1 Rituximab, a chimeric monoclonal antibody (mAb), has been used off-label in the treatment of MS with the phase 2 HERMES study showing reductions in contrast enhancing lesions and the proportion of patients that were relapse free, 2 and the phase 2/3 OLYMPUS study demonstrating further safety and possibly some benefit in decreasing disability progression in primary progressive patients in subgroups that were younger or those with contrast enhancing lesions. 3 Ocrelizumab is a humanized mAb associated with increased antibody-dependent cell-mediated cytotoxic effects and reduced complement-dependent cytotoxic effects as compared to rituximab. 4 As a humanized molecule, ocrelizumab is expected to be less immunogenic with repeated infusions and thus may present a more favourable safety profile when compared to rituximab. 5
The HERMES group showed that a significantly higher number of patients in the rituximab group versus the placebo group had infusion related reaction (IRR) events within 24 hours after the first infusion (78.3% vs 40.0%). However, the rate of IRR after the second dose two weeks later decreased to 20.3%. For all IRRs on rituximab, 92.6% were classified as grade 1 or 2 with the remaining 7.4% being grade 3. Other studies on MS patients treated with rituximab report a 19%-26% rate of IRRs classified as grade 1 or 2, with no IRRs classified as ≥3.6–9
Data from the OPERA I and II and ORATORIO phase 3 trials on ocrelizumab for relapsing and progressive MS patients showed that IRRs were the most common adverse events (AEs). 10 The percentage of relapsing MS patients experiencing IRRs was higher in the ocrelizumab group compared with the interferon β-1a group (34.3% vs. 9.7%) in the OPERA studies 4 and compared to placebo (39.9% vs 25.5%) in ORATORIO. 11 The rate of IRRs was highest during the first infusion and decreased over time with subsequent infusions for the ocrelizumab group. 10 The reported IRRs were primarily mild to moderate in severity in both studies. Severe IRRs for participants treated with ocrelizumab occurred in 2.4% and 1.2% of relapsing and progressive patients, respectively.
With the FDA approval of ocrelizumab in March 2017, patients treated with rituximab were sometimes switched to ocrelizumab for a variety of reasons, such as insurance preference, availability due to FDA approval, and/or increased comfort. The worldwide use of ocrelizumab continues to grow with over 200,000 MS patients treated globally as of April 2021.12,13 Ocrelizumab is the only MS disease modifying agent approved for the treatment of relapsing forms of MS, active secondary progressive MS (SPMS), clinically isolated syndrome (CIS), and primary progressive MS (PPMS) in the United States. Ocrelizumab is approved for active relapsing forms of MS and early PPMS in the European Union (EU). Rituximab continues to remain a first line treatment in Sweden, 14 and it is commonly used off-label in the US to treat MS, although the dosing remains variable. In the United States, rituximab is covered in some states by Medicaid and Medicare as well as some commercial payers including Kaiser Permanente. Immunogenicity results from the OPERA I and II trials showed that only 3 patients (0.4%) showed treatment-induced anti-drug antibodies (ADA) to ocrelizumab. 7 Of these, 1 patient tested positive for neutralizing antibodies (NAB) to ocrelizumab. During the open label extension phase, the prevalence of ADA continued to remain low with post baseline incidence of 1.9% (2/103 with treatment induced ADA). Currently there is little evidence examining the prevalence of treatment-induced ADAs to both rituximab and ocrelizumab in patients that switch from the former.
It is therefore important to demonstrate whether switching from a chimeric anti-CD20 to a fully humanized anti-CD20 leads to an unexpected increase in infusion reactions. Accordingly, we examined IRRs in patients who switched from rituximab to ocrelizumab compared to those continuing on rituximab, specifically to explore the magnitude of the IRRs and subsequent tolerability of ocrelizumab in a real-world population. Factors associated with IRRs were also explored.
Materials and methods
Study population
Patients with relapsing or progressive forms of MS, aged 18–65, were recruited from the Rocky Mountain MS Center at the University of Colorado and who were on rituximab for at least 2 courses given 6 months apart, with the last dose having been administered within 12 months of screening. Participants were enrolled after a clinical decision was made with their provider about whether they would continue on rituximab or switch to ocrelizumab.
Study design
This prospective open-label cohort trial in MS patients (Figure 1) was approved by the Institutional Review board. Participants who switched from rituximab to ocrelizumab (switch group) and those who stayed on rituximab (comparator group) were matched 1:1 on gender and age. See Figure 1 for the schedule of events. Patients in the switch group received the split first dose of ocrelizumab of 300 mg on Day 1 and Day 15, followed by 600 mg at Week 24. Participants in the comparator group continued with standard of care rituximab, which included safety labs and IRR information. Infusion premedication consisted of 50 mg oral diphenhydramine, 1000 mg oral acetaminophen and 100 mg intravenous methylprednisolone, which could be modified at the discretion of the treating neurologist
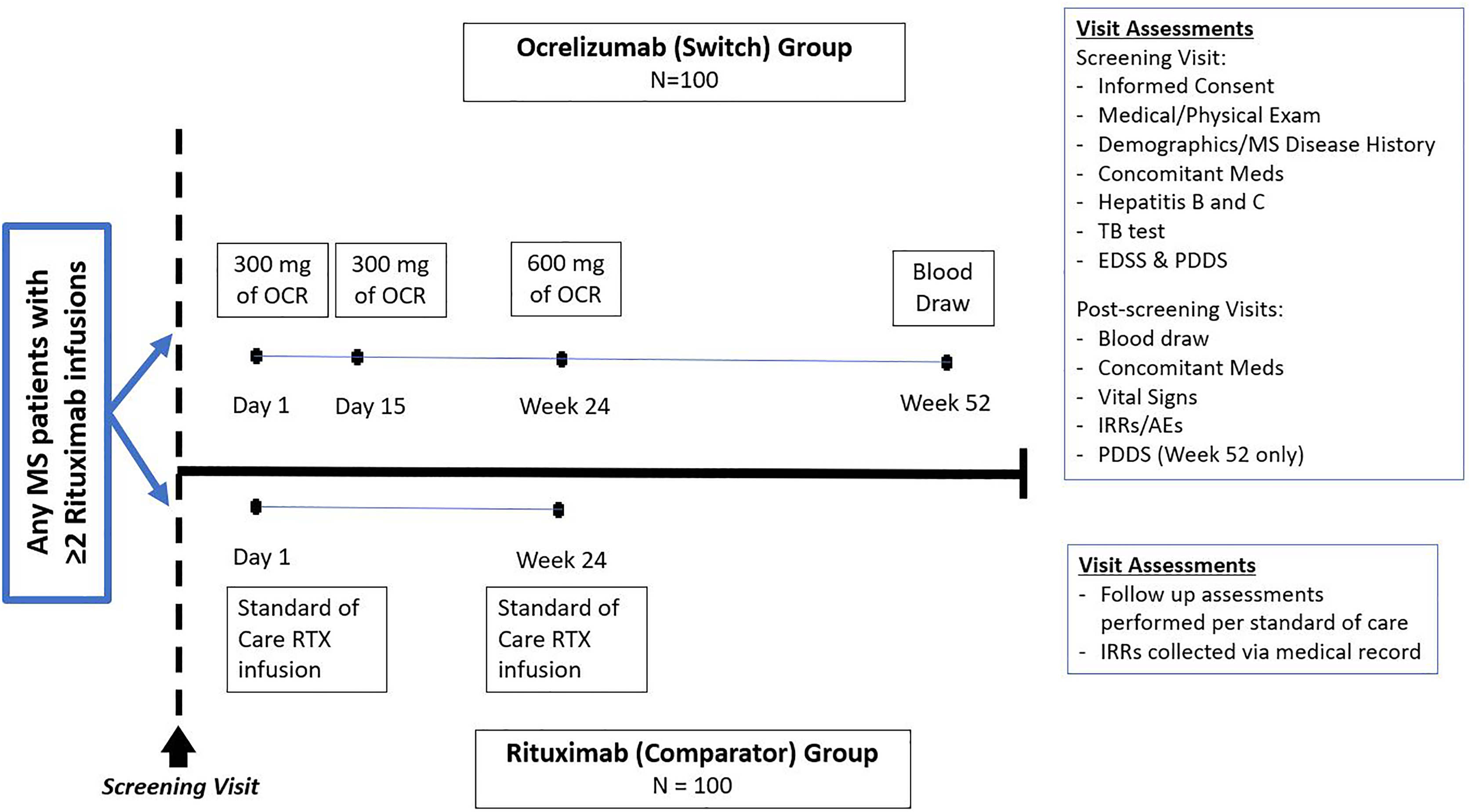
Study design. One hundred patients with multiple sclerosis (MS) were recruited into the switching group to receive ocrelizumab where they were followed for a year and into the comparator group where they continued rituximab and were followed for two infusions. OCR = ocrelizumab, RTX = rituximab, EDSS = Expanded Disability Status Scale, PDDS = Patient Determined Disease Steps, IRR = infusion related reaction, AEs = adverse events.
Infusion related reactions (IRRs)
Following each infusion, nursing staff recorded IRRs using the National Cancer Institute's Common Terminology for Adverse Events Scale: 0 = No infusion reaction; 1 = Mild reaction, infusion interruption not indicated, intervention not indicated; 2 = Requires therapy or infusion interruption but responds promptly to symptomatic treatment; prophylactic medications indicated for 24 hours, 3 = Prolonged, recurrence of symptoms following initial improvement, hospitalization indicated for other clinical sequelae; 4 = Life threatening consequences, urgent intervention indicated; 5 = Death. 15 The post-infusion observation period was 1 hour for both groups. Patient self-assessment of IRRs was also captured.
Laboratory assessments
Safety labs included a negative tuberculosis, Hepatitis B, and C recorded in the patient's chart in the 3 years prior to the screening visit, or it was drawn 30 days prior to infusion. Once enrolled, pre-infusion labs included complete blood count with differential, B and T cell subsets, complete metabolic panel, and an immunoglobulin panel measured at the University of Colorado Hospital Laboratory. Anti-ocrelizumab, and anti-rituximab binding-antibodies were measured in patients who switched to ocrelizumab at baseline and week 24 by PPD Bio A Laboratories (Richmond, VA) and by QPS (Groningen, Netherlands), respectively.
Physical disability assessments
The Expanded Disability Status Scale (EDSS) was used to assess for the disability status of MS patients using unbiased clinical raters in the baseline visit. 16 Additionally, the Patient-Determined Disease Steps (PDDS) was completed by the patient in both study groups and was administered at the screening and termination visits (Week 24). 17
Statistical analysis
Categorical variables were compared between treatment groups with chi-square/Fisher's exact association tests, and continuous variables were compared between treatment groups with T-tests. The 1st and 2nd comparator infusions were similar and combined as one group. Relative risk models were fit for the dichotomized IRR outcomes (present/absent), with the groups: Rituximab, Ocrelizumab Day 1, Ocrelizumab Day 15, and Ocrelizumab Week 24, as the explanatory variables. Generalized estimating equations (GEE), with robust variance estimation, incorporated repeated measures correlation within the same patient. Additional GEE relative risk regression models adjusted for number of previous rituximab infusions (time varying), treatment interaction with log of number of previous rituximab infusions (time varying), baseline age, gender, MS duration, BMI and/or B-cell reconstitution, if either CD19 + and/or CD20 + were ≥1%. Number of previous rituximab infusions was logarithmically transformed because of its skewed distribution, as every patient started with a minimum of two. Significance was set at alpha = 0.05. Tests were performed univariately (without multiple testing adjustment) and in multivariate models. Models were selected with backwards elimination. Statistics were performed in SAS 9.4.
Results
Enrolment took place over 12 months (1/30/17–2/6/18) and details of study participation are shown in Figure 2. The baseline characteristics were similar between the patients who stayed on rituximab (comparator) arm (n = 100) and those that changed their therapy to ocrelizumab (switch) group (n = 100) as seen in Table 1 at the start of the study. The mean MS disease duration was 11.3 years (SD = 7.9). Over 90% of participants had been on other treatments prior to starting rituximab with a mean duration of rituximab of 2.4 years (SD = 1.4) and number of doses of 4.4 (SD = 2.7). Mean time since last rituximab infusion was 219 days (SD = 57) in the switch group and 194 days (SD = 45) in the comparator group (p = 0.0006).

Patient recruitment and enrollment flow chart. For the ocrelizumab (switch) group, 7 patients were lost to follow up at the 12 month visit and lab data was available for only 98 patients on Day 1. n = number.
Patient baseline demographics. n = number, SD = standard deviation, IQR = interquartile range, NA = not available, MS = multiple sclerosis, DMT = disease modifying therapy.

The percent of IRRs in the comparator group (14%), was similar to the switching group on Day 1, (14%, p = 1.00) and on Week 24 (12%, p = 0.65) (Figure 3). Patients in the switch group receiving the second half of the split dose of ocrelizumab on Day 15 had fewer IRRs (4%; p = 0.0075, 0.0209, and 0.0053 for comparison to switching group day 1, switching group week 24, and comparator group respectively). Twenty-one out of 100 patients (21%) in both the switching group and the comparator group had at least one IRR during the 12-month period of the study (p = 1.00). No differences were found in IRRs between comparator and Day 1 or week 24 of the switch group, or between Day 1 and Week 24 within the switching group when further analyses were modeled with covariate adjustments for age, gender, MS disease duration, and BMI. The severity of IRRs were all between 0 (none) and 2 (moderate) with no participants in the comparator or switch groups having an IRR ≥3 that required hospitalization (Supplementary Table 1). Six of the 12 patients who had an infusion reaction at week 24 also had an IRR on Day 1.

Proportion of patients with an infusion related reaction. The relative risks and p values are indicated with significant comparisons shown in red for patients who stayed on rituximab (comparator group) or those who switched to ocrelizumab.
For patients in the switch group, safety monitoring bloodwork is summarized in Table 2. the proportion of patients with IgG levels ≤500 mg/dL did not appreciably change during the study period: 7.1% on day 1, 6.0% on week 24 and 5.5% at month 12. The proportion of switching patients with CD19 counts ≤1% increased from 57.1% on Day 1 to 92.0% at Week 24 and 90.3% at 12 months. Similarly, the proportion of switching patients with CD20 counts ≤1% increased from 56.7% on Day 1 to 92.0% at Week 24 and 88.6% at 12 months. This was associated with a longer mean time since last infusion in the switch group on day 1 of 219 days (SD = 57) versus the expected 180 days if infused every 6 months. There were no differences from Day 1 to week 52 in mean IgA, IgG or IgM. However due to the right skew in the data, the geometric (transformed) means of IgA and IgM decreased statistically significantly from Day 1 to Week 52 by 3.2% [(0.6, 5.8); p = 0.012] and 5.8% [(2.7, 8.9); p < 0.001], respectively. The geometric mean of IgG had a non-statistically significant drop of 5.3% [(-1.7, 11.7); p = 0.171].
Comparison of safety laboratories at Day 1, week 24, and week 52. Units for each row are listed only for the reference range column. SD = standard deviation.

We examined factors associated with IRRs (Table 3) on Day 1 in the switching group. Univariately, the risk of an IRR decreased by 7.1% [risk ratio = 0.929; (0.856, 1.008); p = 0.044] per year of MS duration. The risk of IRR was 26.83% in patients having evidence of B cell reconstitution (defined as having CD19 and/or CD20 counts ≥1%), compared to only 5.36% of the non-reconstituted patients [risk ratio = 5.0081; (1.491, 16.818); p = 0.007]. In a multivariate model, B cell reconstitution, adjusting for the number of the log of the prior rituximab infusions, time since last rituximab infusion, age, gender, MS disease duration, and BMI, increased the risk of IRR by 287.2% [risk ratio = 3.872, (1.208, 12.404) p value = 0.0198]. Backward elimination, starting from the full model, eliminated all explanatory variables for an increased the risk of IRR except B cell reconstitution (Table 3). The same analysis could not be applied to Week 24 of the switching group because there were too few B cell reconstituted patients (n = 8). However, at Week 24 for the 12 patients with an IRR, 6 had an IRR at Day 1 (Kendal Tau correlation = 0.379, p = 0.0015). B cell data was not available for the comparator group.
Factors influencing the risk of IRRs on Day 1. Univariate, multivariate, and multivariate analysis with backward elimination models are presented. *The only factor that remained in the multivariate analysis with backward elimination was B cell reconstitution CD19 and/or CD20

Only one patient was found to have antidrug antibodies to ocrelizumab. These antibodies were already present on Day 1 prior to ocrelizumab exposure and were still present at week 24. This patient did not have an infusion reaction and was CD19 and CD20 depleted (<1%). Six additional patients had a positive test on screening, and only one of these patients had an IRR. Anti-ocrelizumab antibodies were not associated with IRR (p = 1.000), or with CD19 or CD20 repletion (p = 0.233 and 0.135, respectively).
Of the 100 participants who switched to ocrelizumab, 16 had ADA to rituximab at Day 1, which decreased to 7 participants by week 24 with 5 having them at both time points (p value of McNemar test = 0.0386). An increased number of rituximab infusions prior to switching to ocrelizumab (log scale) was associated with a decreased chance of rituximab antibodies [risk ratio = 0.250 per doubling of rituximab infusions, (0.069, 0.909); p = 0.003]. Each increase by 7 days since the last rituximab infusion was associated with a decrease in the chance of screening anti-ocrelizumab antibodies by 21.8% [(4.7, 35.9); p = 0.026].
Adverse events (AEs) besides IRRs reported during the study are presented in Supplementary Table 2. The most common AEs experienced by participants in this trial were upper respiratory infections, headache/migraine, nausea, numbness in extremities, and fatigue. No Serious AEs (SAEs) were reported, and no patients dropped out due to an adverse drug experience.
Discussion
Rituximab has been used as an off-label treatment for MS in some MS centers since completion of the HERMES and Olympus trials. Since approval of ocrelizumab in March of 2017, it has become the most commonly prescribed treatment for MS in patients starting a new treatment in the United States. 13 Switching between these two anti-CD20 mAbs has become commonplace as patients change insurance. Here we show that safety and tolerability are comparable when switching from rituximab to ocrelizumab.
Our results demonstrate that there is no increase in frequency or severity of IRRs when switching from rituximab to ocrelizumab. The IRR rate and severity with the first 300 mg dose of ocrelizumab (14%) or 600 mg at 24 weeks (12%) was similar to those in patients who continued on to received rituximab infusions (14%). These patients who had prior anti-CD20 exposure had a lower rate of IRRs than treatment naïve patients receiving the first 300 mg dose of ocrelizumab in the OPERA studies where 27.5% of subjects had an IRR. 4 Similar to the current study, most IRRs in the OPERA trials were evaluated to be mild to moderate and occurred mainly with the first dose of ocrelizumab. In OPERA, 1.7% of patients had severe reactions with one life-threatening IRR, which we did not observe in this study, but were not powered to detect. The overall IRR rates were similar to those seen in other studies such as ORATORIO for progressive MS patients 10 (27.4% for the first infusion) and ENSEMBL PLUS 18 (23.1% in the conventional infusion and 24.6% in the shorter infusions at Day 1). The rate of IRRs in subsequent infusions in more observational (and less controlled) trials for a rapid infusion protocol are more variable ranging from 12.4% in the CHORDS study to 48.4% in the SaROD study. 19 However, the frequency and length of follow up differed quite substantially between these trials such at the reporting probably was higher in OPERA where participants were followed every 4 weeks via phone along with an ascertainment bias in studies specifically designed to look at IRRs and especially those that were open label such as SaROD. These results suggest that there does not appear to be a difference in IRR rate or severity between a chimeric anti-CD20 mAb versus a humanized mAb with increased antibody-dependent cell-mediated cytotoxic activity.
This is a pragmatic study and was limited by the nature of the comparator group, as we utilized a sample of convenience. Although we did not have any IRRs rated above 2, it is still a relatively small sample and thus does not rule out the possibility of more severe IRRs occurring on rare occasions for patients on anti-CD20 mAbs. Additionally, as we enrolled participants after the decision to either remain on rituximab or switch to ocrelizumab was made, which could include some selection bias as the patients who switched may have had more reactions on rituximab, for example. The main reason, however, for switching was due to insurance changes due to the FDA approval of ocrelizumab. Measurement bias may have resulted in differences on IRR, as the rituximab patients received their infusions as standard of care while ocrelizumab patients in the switch arm were infused in a research unit. To help minimize this, a training was conducted for infusion nurses at both locations on measuring IRRs. ADAs were evaluated in this study but were not found to be associated with IRRs. Seven patients had anti-ocrelizumab antibodies on screening and only 1 was confirmed to have these antibodies. This patient did not have an IRR and was still B-cell depleted making the significance of these antibodies unknown. These antibodies may be associated with serum sickness, 20 but the lack of a commercial test limits the ability to assess this for ocrelizumab. However, these antibodies have been associated with serum sickness in rituximab, 21 which can be tested commercially. Serum sickness is an immune-complex mediated hypersensitivity reaction that occurs 7–10 days after an infusion. Antibodies to rituximab, as expected, were not found to be associated with IRRs to ocrelizumab but were also not found to be associated with B cell reconstitution. However, patients who had longer exposure to rituximab were found to have a higher likelihood of having anti-rituximab antibodies. Lower levels of rituximab have been previously associated with the formation of these antibodies in rheumatoid arthritis patients, which did not affect clinical outcomes. 22
The only factor that did associate with having IRRs was the presence of B-cells. This is suggested by the much lower rate of IRR in the second dose of the initial course of infusions with ocrelizumab seen in this study (4%), as well as in OPERA (4.7%) and ORATORIO (7.3%). 10 We could explore this further on Day 1 of this study, as some participants had a delay in getting their infusions and began to have B-cell reconstitutions. In a multivariate analysis, the only factor associated with IRRs was found to be a CD19 and/or CD20 count ≥1%. At subsequent infusions, IRRs were associated with having had a prior IRR.
In this study, we report good safety and tolerability in switching from rituximab to ocrelizumab. No new safety concerns were identified in this study and IRRs were similar when switching treatments in patients with MS. Many patients with IRRs will tend to push off infusions but this study suggests that patients with IRRs should be infused on a more regular and possibly frequent schedule to minimize the chance of reconstitution. Specifically, patients switching from rituximab to ocrelizumab should be infused around 6 months from their last infusion or at least not have their infusions extended to minimize IRRs if their B-cells are reconstituting prior to their next infusions.
Supplemental Material
sj-docx-1-mso-10.1177_20552173211069359 - Supplemental material for Tolerability and Safety of Switching from Rituximab to Ocrelizumab: Evaluating Factors Associated with Infusion Related Reactions
Supplemental material, sj-docx-1-mso-10.1177_20552173211069359 for Tolerability and Safety of Switching from Rituximab to Ocrelizumab: Evaluating Factors Associated with Infusion Related Reactions by Enrique Alvarez, Kavita V. Nair, Stefan Sillau, Ian Shelton, Rebecca Seale, Sean Selva, John Corboy and Timothy L Vollmer in Multiple Sclerosis Journal – Experimental, Translational and Clinical
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Enrique Alvarez, MD has received compensation for activities such as advisory boards, lectures and consultancy with the following companies and organizations: Actelion/Janssen, Bayer, Biogen, Celgene, EMD Serono, Genentech, Genzyme, Novartis, and TG Therapeutics. He has received research support from the following: Biogen, Genentech, Novartis, TG Therapeutics, Patient Centered Outcomes Research Initiative (PCORI) and Rocky Mountain MS Center. Kavita V Nair, PhD consults for Bristol Meyers Squibb, Novartis, Genentech, Biogen and EMD Serono and has grants from Genentech, Novartis and Bristol Meyers Squibb. John Corboy, MD has received compensation for activities such as advisory boards, lectures and consultancy with the following companies and organizations: Mylan, Novartis, Prime CME, Rocky Mountain MS. He has received research support from the following: MedDay, Novartis, National MS Society, Patient Centered Outcomes Research Initiative (PCORI). Timothy Vollmer, MD has received compensation for lectures and consultancy with the following: Biogen IDEC, Genentech/Roche, Siranax, Celgene, EMD Serono and Novartis. He has received research support from the following: Rocky Mountain MS Center; Biogen; Actelion; Roche/Genentech; F. Hoffman-La Roche, Ltd and TG Therapeutics, Inc. The remaining authors declare that they have no conflicts of interest.
Funding
This study was an investigator sponsored trial funded by Genentech.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.