
Abstract
Background
Clinical phenotypes of patients with antibodies to myelin oligodendrocyte glycoprotein (anti-MOG+) are unknown in India.
Objectives
Retrospectively to characterise anti-MOG+ patients with inflammatory central nervous system disorders in India.
Method
A total of 87 patients with non-multiple sclerosis demyelinating disorders (excluding acute disseminated encephalomyelitis) who were seronegative for anti-aquaporin 4 antibody were retrospectively analysed using a cell-based assay for anti-MOG+ status.
Results
Twenty-five patients were anti-MOG+ in this cohort. They represented 28.7% (25/87) of patients who tested negative for anti-AQP4+. Sixty-four per cent (16/25) of anti-MOG+ patients were men and had a relapsing course. Patients with recurrent optic neuritis and those with a single attack of acute longitudinally extensive transverse myelitis were the most common phenotypes.
Conclusion
Relapsing optic neuritis was the most common phenotype, contrasting with a lower risk of relapses in transverse myelitis.
Introduction
A significant proportion of non-Caucasian patients with idiopathic inflammatory disorders have atypical features for multiple sclerosis (MS). Among these, patients with autoantibodies against myelin oligodendrocyte glycoprotein (anti-MOG+) have been described in those who were negative for aquaporin-4 antibodies (anti-AQP4).1–7 In this study we describe the frequency and clinical characteristics of anti-MOG+ patients among Indian patients.
Patients and methods
All patients were obtained from the Mangalore demyelinating disease registry that has prospectively enrolled all consecutive patients with central nervous system (CNS) demyelinating disorders on the southwestern coast of India. The inclusion criteria for this study were as follows: (1) a clinical diagnosis of ‘non-MS’ CNS inflammatory demyelinating disorders including monophasic and recurrent disorders; (2) seronegative status for AQP4+ by live cell-based assay. Patients presenting with acute disseminated encephalomyelitis (ADEM) were not included. Anti-AQP4 and anti-MOG cell-based assay was blindly performed at Tohoku University using live transfected HEK-293 cells with AQP4-M23 isoform and full-length MOG as described previously. 3 All patients had blood draws during an attack and prior to initiating parenteral steroids. In 64% of patients with relapsing disease the samples were obtained during the first relapse. Clinical and magnetic resonance imaging (MRI) data were reviewed retrospectively. This study was approved by the institutional ethics committee and all patients signed an informed consent form.
Results
Anti-MOG+ patients – clinical course, MRI and therapy.

anti-MOG+: autoantibodies against myelin oligodendrocyte glycoprotein; MRI: magnetic resonance imaging; NMO: neuromyelitis optica; RTM: recurrent transverse myelitis; ATM: acute transverse myelitis; RON: recurrent optic neuritis; AZA: azathioprine; MMF: mycophenolate mofetil; VFS: visual functional score; EDSS: Expanded Disability Status Scale; NT: not treated; NA: not applicable.
Anti-MOG antibody negative patients (n=62) included isolated LETM 45.2% (28/62), NMO 27.4% (17/62), RON 17.7% (11/62) and recurrent LETM 9.7% (6/62). Seronegative patients had a similar age of onset of disease as anti-MOG+ patients (see Supplementary Tables 1 and 2), no gender predilection and had greater disability. The latter may be accounted for by the high number of patients with LETM in this group.
Unilateral or bilateral thickening of the optic nerve with T2 weighted (T2W) hyperintense signals and/or patchy enhancement was seen in 61.5% (8/13) of anti-MOG+ patients. Optic chiasm was partially involved (Figure 1(a–c)) in 15.4% (2/13). The dorsal cord was involved in 57.1% (8/14) of patients (Figure 1(d)) presenting with long segment myelitis. In the remainder, cervical cord lesions remained contained and did not extend into the cervicomedullary or caudal brainstem regions. Axial sections showed involvement of the central part of the cord in 64.3% (9/14) who had myelitis. The most common abnormality on brain MRI was subcortical white matter lesions (atypical for MS), which was seen in 24% (6/25) of patients (Figure 1(e and f)). Two patients (8%) with brain lesions had altered sensorium and headache at onset. There were no striking differences in imaging between anti-MOG+ and seronegative patients.
Magnetic resonance images of the spinal cord and optic nerve in anti-MOG+ patients. Fat suppressed (FATSAT) axial image of the orbit (a) and T2 weighted (T2W) coronal images in a 31-year-old woman with recurrent optic neuritis showing thickened and hyperintense intraorbital (a) and intracranial segment (b) of the left optic nerve with extension into the optic chiasm on the left side (c). (d) T2W sagittal image of the spinal cord showing hyperintense linear cord lesion extending from upper dorsal cord to the conus in a 21-year-old man with isolated transverse myelitis. (e) and (f) 25-Year-old woman with recurrent myelitis showing atypical brain lesions.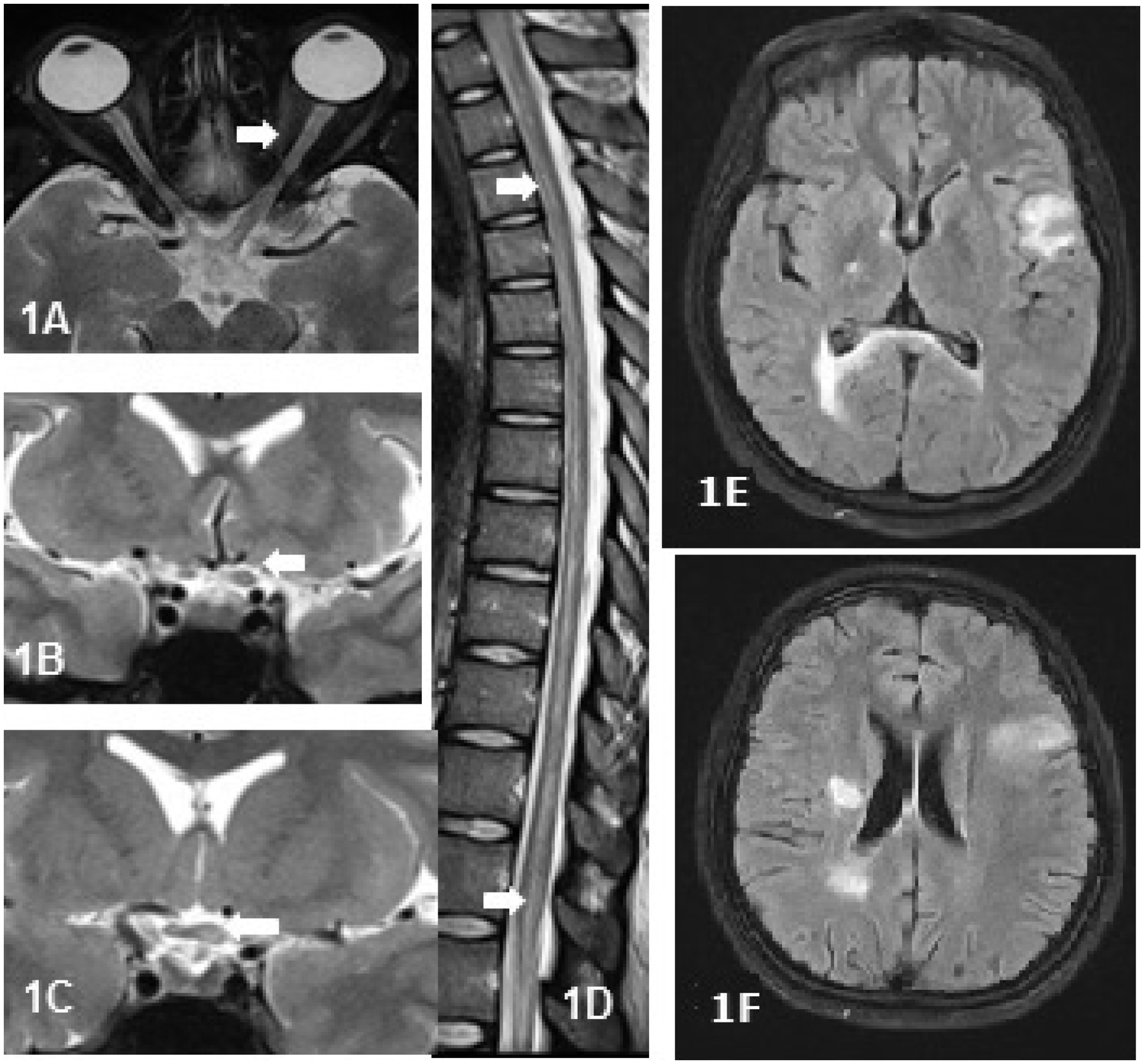
In this retrospective analysis, all relapsing anti-MOG+ patients had been treated with immunosuppressant therapy (Table 1). Two patients with anti-MOG+ RON remained free of new attacks after discontinuation of immunosuppressants (median follow-up of 22 months). Three patients with RON received steroids during relapses but they did not take long-term immunosuppressants (Table 1; patients 17, 20, 25). One patient had been treated elsewhere initially with beta-interferon for 12 months, with clinical worsening.
Discussion
We identified a significant subgroup of patients (28.7%) with anti-MOG+ among patients with idiopathic inflammatory CNS disorders who were negative for anti-AQP4+. Relapsing optic neuritis was the most common phenotype and has been reported previously.9,10 But patients with a single attack of LETM affecting the dorsal cord were also frequent. The reason for a higher risk of relapses in patients with optic neuritis is unknown. Further prospective longitudinal studies may clarify whether those patients maintain anti-MOG+ for long periods. These patients shared some clinical similarities with anti-AQP4+ NMO spectrum disorders with limited phenotypes such as RON and LETM. NMO, satisfying Wingerchuck, 2006 criteria 8 was seen in two patients only (cases 1 and 2, Table 1). Symptomatic brainstem lesions causing nausea, vomiting and hiccups were uncommon. Thus the majority of patients did not fulfill the new diagnostic criteria for NMO spectrum disorders. 11
The visual and disability outcome in our cohort after more than 5 years of disease was good in the majority of patients, but anti-MOG+ disease may occasionally be very severe as was evident in one of our patients. The MRI of the spinal cord showed a predilection for dorsal cord involvement and a lack of cranial extension of cervical cord lesions into the brainstem commonly seen in anti-AQP4+ NMO spectrum disorders. Bilateral simultaneous optic neuritis was uncommon in our cohort, but the orbital MRI showed bilateral long segment optic nerve involvement in those bilaterally affected with optic neuritis. Unlike other studies3,7,12 we found partial involvement of the optic chiasm on the orbital MRI of two patients with RON. First line disease-modifying drugs used for MS such as beta-interferon may worsen anti-MOG+ diseases as experienced by one of our patients. Several reports showed an increase in relapses, worsening of disability, 13 and an increase in the NMO-IgG titre 14 while on disease-modifying therapy with beta interferon. This may be due to a difference in immune pathogenesis between MS and antibody-mediated disorders such as anti-AQP4+ and anti-MOG + disorders. Response to primary immunosuppressant therapy in our small cohort of relapsing anti-MOG+ patients was satisfactory, with none of our treatment-compliant patients experiencing breakthrough relapses.
In conclusion, almost a third of our cohort of patients from India with non-MS demyelinating disorders and negative for anti-AQP4+ had anti-MOG positivity. Relapsing optic neuritis was the most common phenotype, contrasting with those patients who had a single attack of LETM. Long-term immunosuppressive therapy may benefit those patients with a relapsing disease course. A significant number of patients in our cohort were seronegative underscoring the heterogeneity in aetiopathogenesis of these disorders. A prospective study that includes a larger cohort and a wider spectrum of disorders such as MS and ADEM may overcome some of the limitations of this study.
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Lekha Pandit, Sharik Mustafa, Anitha D’Cunha, Chaithra Malli, Akshatha Sudhir and Toshiyuki Takahashi. Dr Sato has received speaker honoraria from Novartis. Dr Fujihara serves on scientific advisory boards for Bayer Schering Pharma, Biogen Idec, Mitsubishi Tanabe Pharma Corporation, Novartis Pharma, Chugai Pharmaceutical, Ono Pharmaceutical, Nihon Pharmaceutical, Merck Serono, Alexion Pharmaceuticals, Medimmune and Medical Review; has received funding for travel and speaker honoraria from Bayer Schering Pharma, Biogen Idec, Eisai Inc., Mitsubishi Tanabe Pharma Corporation, Novartis Pharma, Astellas Pharma Inc., Takeda Pharmaceutical Company Limited, Asahi Kasei Medical Co., Daiichi Sankyo, and Nihon Pharmaceutical; serves as an editorial board member of Clinical and Experimental Neuroimmunology (2009 to present) and an advisory board member of the Sri Lanka Journal of Neurology; has received research support from Bayer Schering Pharma, Biogen Idec Japan, Asahi Kasei Medical, The Chemo-Sero-Therapeutic Research Institute, Teva Pharmaceutical, Mitsubishi Tanabe Pharma, Teijin Pharma, Chugai Pharmaceutical, Ono Pharmaceutical, Nihon Pharmaceutical, and Genzyme Japan.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Dr Sato has received scholarship from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, grant-in-aid for scientific research from the Japan Society for the Promotion of Science (KAKENHI 15K19472), research support from CAPES/Brasil (CSF-PAJT – 88887.091277/2014-00). Dr Fujihara is funded as the secondary investigator (grant number 22229008, 2010–2015) by the grants-in-aid for scientific research from the Ministry of Education, Science and Technology of Japan and as the secondary investigator by the grants-in-aid for scientific research from the Ministry of Health, Welfare and Labor of Japan (2010 to present).