
Abstract
Background
Optic neuritis (ON) occurs in immune-mediated disorders including multiple sclerosis (MS), aquaporin-4 antibody-positive (AQP4) neuromyelitis optica spectrum disorder (AQP4-NMOSD) and myelin oligodendrocyte glycoprotein (MOG) antibody-associated demyelination (MOGAD). Accurate determination of aetiology is critical for appropriate treatment and prognostication.
Objective
To evaluate demyelination and axonal loss in MOG-ON to facilitate differentiation from MS-ON and AQP4-ON.
Methods
15 MOGAD patients with previous ON (25 eyes) underwent multifocal visual evoked potential (mfVEP) recordings and optical coherence tomography scans. Comparison was made to previously reported MS patients (n = 67, 69 eyes) and AQP4-NMOSD patients (n = 15, 23 eyes) with prior ON and healthy controls (n = 37, 74 eyes).
Results
MOG-ON patients had less retinal nerve fibre layer (RNFL) loss than AQP4-ON patients (p < 0.05) and less mfVEP latency prolongation than MS-ON patients (p < 0.01). Number of ON episodes in MOGAD was associated with reduced RNFL thickness (global, p = 0.07; temporal, p < 0.001) and mfVEP amplitude (p < 0.001). There was no abnormality in non-ON eyes.
Conclusions
Our study demonstrated a distinct pattern of damage in MOG-ON compared to AQP4-ON and MS-ON. ON in MOGAD produces less axonal loss than AQP4-NMOSD. Damage accumulates with relapses, supporting the role of maintenance immunosuppression to induce remission. Compared to MS, MOGAD causes less demyelination.
Keywords
Optic neuritis (ON) is characterised by visual loss, pain on eye movements and impaired colour vision due to optic nerve inflammation. It occurs as a manifestation of immune-mediated disorders including multiple sclerosis (MS), aquaporin-4 (AQP4) antibody-positive neuromyelitis optica spectrum disorder (AQP4-NMOSD) and myelin oligodendrocyte glycoprotein (MOG) antibody-associated demyelination (MOGAD). The latter two disorders are associated with antibodies targeting AQP4, a water channel protein, and MOG, a myelin sheath protein, respectively. ON is seen frequently in all three disorders and may relapse.
Clinical and radiological features may suggest the underlying cause of ON. Simultaneous bilateral ON with longitudinally extensive involvement of the optic nerve is present in both AQP4-ON and MOG-ON.1,2 MOG-ON is associated with more anterior visual pathway involvement including optic disc swelling, as well as prominent optic nerve oedema and perineural enhancement on imaging, and rapid steroid responsiveness.2–4 AQP4-ON tends involve the posterior optic pathways, including the chiasm and optic tracts, and has a particular predilection for the hypothalamus. 2 MS-ON is more often unilateral with shorter segment optic nerve involvement and is associated with typical lesions at characteristic locations throughout the neuraxis. 2
Accurate determination of the aetiology of ON is critical because this has implications for treatment decisions and assessment of visual prognosis. Delayed diagnosis and immunotherapy in antibody-associated demyelination can result in sustained visual impairment.3–5 Immunomodulatory therapies which are highly successful in MS can cause deterioration in AQP4-NMOSD and MOGAD, whereas remission can be achieved with long term immunosuppression.1,6–9 Using electrophysiological parameters (multifocal visual evoked potentials (mfVEPs)) in combination with clinical and radiological measures may facilitate this diagnostic decision making.
Neurodegenerative changes in optic pathways have been demonstrated following ON in MS, AQP4-NMOSD and MOGAD. Objective quantification of the degree of axonal loss using optic coherence tomography (OCT) has shown irreversible damage to the optic nerve in all three disorders.5,10 Visual evoked potentials (VEPs) are used clinically to demonstrate demyelination in addition to axonal injury in the optic nerve.10,11 More accurate and sensitive topographic functional evaluation of the visual pathway is achieved by simultaneous recording across multiple segments of the visual field using mfVEPs. 12 This modality has not yet been assessed in MOG-ON. In this study we aimed to evaluate markers of demyelination and axonal loss in MOG-ON compared to AQP4-ON and MS-ON, to identify patterns of abnormality which can facilitate clinical differentiation between these three conditions.
Materials and Methods
Participants
MOGAD patients with previous ON (n = 15, 25 eyes) were identified using a live cell-based assay analysed by flow cytometry and individually recruited.3,13 Consecutively recruited MS (n = 67, 69 eyes) and AQP4-NMOSD (n = 15, 23 eyes) patients with previous ON and healthy control subjects (n = 37, 74 eyes) with similar gender and age distributions were reported previously. 14 MS was diagnosed according to the 2010 revised McDonald criteria 15 and AQP4-NMOSD was diagnosed according to the 2015 diagnostic criteria. 16 Non-ON eyes from each group were analysed separately. Exclusion criteria included retinal, optic nerve or other neurologic diseases affecting the visual system including any other types of optic neuropathy and optic atrophy without known cause. Patients in the 12 months following acute ON were excluded to avoid the amplitude reduction and latency prolongation in mfVEPs in post-acute ON. 17 Patients with severe visual loss (visual acuity ≥1 logMAR) were unable to perform mfVEPs and were therefore excluded. This research study was conducted in accordance with the principles of the Declaration of Helsinki and was approved by the Human Research Ethics Committee of the University of Sydney (Sydney, Australia) [2013/106]. Written informed consent was obtained from all participants.
OCT scans and imaging analysis
All participants underwent an OCT scan using Heidelberg Spectralis OCT and Eye Explorer software version 6.9.5.0 (Heidelberg Engineering, Heidelberg, Germany) as described previously18–20 on the same day as other tests. A peripapillary ring scan (diameter, 3.50 mm; manual placement of ring) was performed to obtain retinal nerve fibre layer (RNFL) thickness measurements for the global RNFL and temporal RNFL sector with automated segmentation. The pupils of participants were not dilated before examination. All tests were performed at a single site with the same operator present and device under room light conditions. Image quality was assessed using the OSCAR-IB criteria.21 There was no post-acquisition discard or post-processing of images. OCT data presented in this study were in line with international criteria. 22
mfVEP recordings
mfVEP testing was performed using Vision Search 1 (VisionSearch, Sydney, Australia) controlled by Terra software (VisionSearch), and standard stimulus conditions as described previously. 23 56 closely packed segments (eccentricity up to 24°) in a cortically scaled dart-board configuration were used. Each segment contained 16 black-and-white checks, which reversed patterns according to a pseudorandom sequence. Four gold-disc electrodes (Grass, West Warwick, RI) were used for bipolar recording with two electrodes positioned 4 cm on either side of the inion, one electrode 2.5 cm above and another 4.5 cm below the inion in the midline. Electrical signals were recorded along two channels, demonstrating the difference between superior and inferior, as well as between left and right electrodes.
The largest peak-to-trough amplitude within the interval of 70 to 210 ms was established for each channel. The wave of maximum amplitude among the channels was selected automatically by software to create a combined topographic map for amplitude and latency analysis. Traces from the same channel were selected for all tests for each individual segment of the visual field by a specially designed algorithm. 24
Statistical analysis
Statistical analysis was conducted using SPSS software version 25 (SPSS, Inc. Chicago, IL). Data from ON eyes only were included for comparison between groups using the generalized estimating equation (GEE) model. Data were presented as the mean ± standard deviation. Visual acuity was expressed as logarithm of the minimum angle of resolution values. Standard values were used, with counting fingers, 1.7; hand movements, 2.0; no light perception, 3.0. 25 The GEE model was also used for the multiple group parametric comparison. Age, gender, number of ON episodes and affected eye (intra-subject factor) were included as covariates in the GEE model analysis. P values less than 0.05 were considered statistically significant. A Bonferroni correction was not required because a small number of pre-planned analyses were performed. 26
Results
The demographic data and clinical features of the included patients are presented in Table 1. Comparison was made to previously reported AQP4-ON and MS-ON patients. 14 Dedicated orbital MR imaging contemporaneous to the first episode of optic neuritis was available for a subset of patients in the study and included in Supplementary Table 1. MOG antibody binding ratios are presented in Supplementary Table 2.27,28 There was a trend towards more recurrent ON in the MOG-ON cohort than the MS-ON cohort (p = 0.06) but no difference compared to the AQP4-ON cohort. Notably there were two MOG-ON patients who reported more than 20 episodes affecting each eye. Visual acuity was significantly more impaired in the AQP4-ON cohort than the MOG-ON cohort. There were no other significant differences between groups.
Demographic data and clinical features of MOG-ON, AQP4-ON and MS-ON patients.
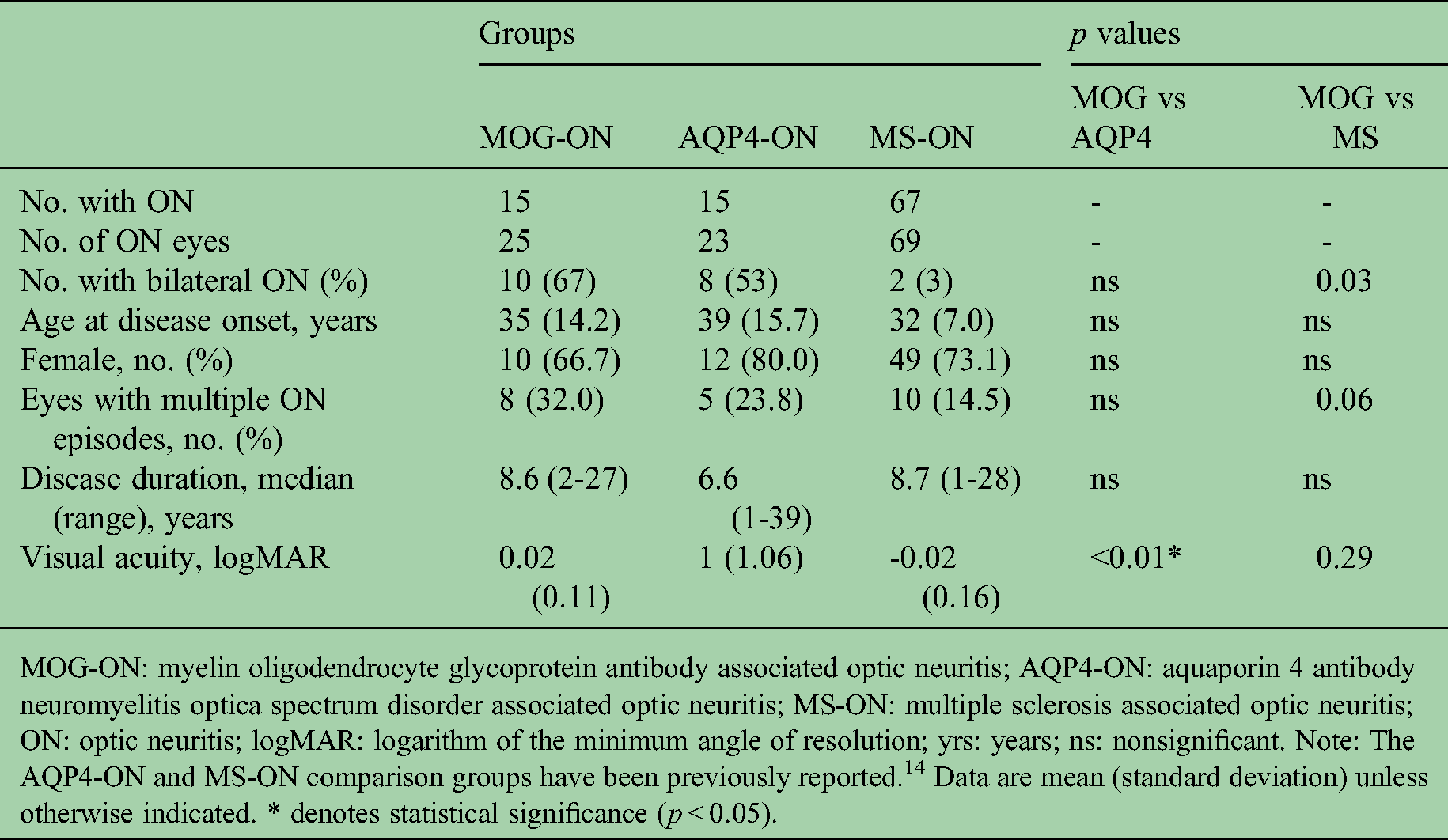
MOG-ON: myelin oligodendrocyte glycoprotein antibody associated optic neuritis; AQP4-ON: aquaporin 4 antibody neuromyelitis optica spectrum disorder associated optic neuritis; MS-ON: multiple sclerosis associated optic neuritis; ON: optic neuritis; logMAR: logarithm of the minimum angle of resolution; yrs: years; ns: nonsignificant. Note: The AQP4-ON and MS-ON comparison groups have been previously reported. 14 Data are mean (standard deviation) unless otherwise indicated. * denotes statistical significance (p < 0.05).
The global and temporal RNFL thickness, mfVEP amplitude and latency for the MOG-ON, AQP4-ON and MS-ON groups are presented in Table 2. There was a significantly greater reduction in global RNFL thickness in AQP4-ON (62.7 ± 22.3 µm) compared to MOG-ON (69.0 ± 23.1 µm) (p < 0.05) and a trend towards significance for temporal RNFL thickness (44.0 ± 16.6 µm vs 48.3 ± 14.9 µm) (p = 0.07) (Figure 1). There was also a trend towards greater reduction of mfVEP amplitude in the AQP4-ON group (129.9 ± 51.1 µV) compared to MOG-ON (108.1 ± 58.6 µV) (p = 0.06) (Figure 2A). There was a significantly greater prolongation of mfVEP latency in MS-ON (163.2 ± 14.6 ms) compared to MOG-ON (155.2 ± 10.6 ms) (p < 0.01) (Figure 2B), however there was no difference in other measures.

Representative OCT peripapillary ring scans showing RNFL thickness in MOG-ON, AQP4-ON and MS-ON. (A) OCT in an ON eye in a MOGAD subject. (B) OCT in an eye with relapsing ON (>20 episodes) in a MOGAD subject. (C) OCT in an ON eye in a MS subject. (D) OCT in an ON eye in an AQP4-NMOSD subject. Note: Subjects C and D were part of a previously reported cohort. 14

Comparison of multifocal visual evoked potential (mfVEP) measures in MOG-antibody associated demyelination (MOGAD), AQP4-positive neuromyelitis optica spectrum disorder (AQP4-NMOSD) and multiple sclerosis (MS) patients. A. mfVEP amplitude measures in MOGAD, AQP4-NMOSD and MS patients. B. mfVEP latency measures in MOGAD, AQP4-NMOSD and MS patients. Error bars represent standard deviation. Note: The AQP4-ON and MS-ON comparison groups have been previously reported. 14 Statistical significance is indicated by *p < 0.05 and **p < 0.001.
Electrophysiological and OCT parameters in ON eyes.

gRNFL: global retinal nerve fibre layer thickness; tRNFL: temporal retinal nerve fibre layer thickness; mfVEP: multifocal visual evoked potential; MOG-ON: myelin oligodendrocyte glycoprotein antibody associated optic neuritis; AQP4-ON: aquaporin 4 antibody neuromyelitis optica spectrum disorder associated optic neuritis; MS-ON: multiple sclerosis associated optic neuritis. Note: The AQP4-ON and MS-ON comparison groups have been previously reported. 14 Data are mean (standard deviation) unless otherwise indicated. * denotes statistical significance (p < 0.05).
The number of MOG-ON episodes was associated with reduction in both global and temporal RNFL thickness (p = 0.07 and p < 0.001 respectively) and mfVEP amplitude (p < 0.001) (Figure 3). There was a non-significant relationship to mfVEP latency (p = 0.47) and visual acuity (p = 0.43). In AQP4-ON, there was a trend towards reduced global and temporal RNFL thickness (p = 0.06 and p = 0.09 respectively) with relapsing ON, but not mfVEP amplitude (p = 0.51), VEP latency (p = 0.41) or visual acuity (p = 0.59). In the MS cohort, there was no correlation between number of ON episodes and these parameters.

Changes in visual acuity, retinal nerve fibre layer (RNFL) thickness and multifocal visual evoked potential (mfVEP) measures with relapsing optic neuritis (ON) in MOG-antibody associated demyelination (MOGAD). (A) Effect of increasing number of ON episodes on visual acuity. (B) Effect of increasing number of ON episodes on RNFL thickness. (C) Effect of increasing number of ON episodes on mfVEP amplitude and latency.
In non-ON eyes in the MOGAD cohort, there was no RNFL loss (gRNFL 96 ± 12.88 µm and tRNFL 66.8 ± 8.56 µm), reduction in mfVEP amplitude (170.40 ± 57.84 µV) or prolongation of mfVEP latency (145.60 ± 3.85 ms) compared to controls. Further analysis could not be performed due to small numbers of non-ON eyes in this cohort.
Discussion
This study characterises structural and functional markers of optic nerve dysfunction in a cohort of MOGAD patients with ON. Our data demonstrates a distinct pattern of clinical, retinal imaging and electrophysiological features in MOG-ON compared to AQP4-ON and MS-ON. Overall, MOG-ON results in less severe axonal loss than AQP4-ON, and a similar degree of axonal loss to MS-ON, reflected in a lesser degree of global RFNL thinning and greater preservation of visual acuity in the chronic post-ON period. There is also a trend towards higher mfVEP amplitude in the MOG-ON group. However, recurrent episodes of ON result in gradual accumulation of axonal loss with each episode of demyelination. This is demonstrated by greater loss of both global and temporal RNFL thickness and reduced mfVEP amplitude. Compared to MS-ON, MOG-ON produces less demyelination with significantly less prolongation of mfVEP latency. The degree of demyelination also does not accumulate with recurrent episodes of MOG-ON.
Previous OCT studies have demonstrated greater thinning of RNFL and macular ganglion cell and inner plexiform layer in AQP4-ON and MOG-ON than MS-ON, providing evidence of irreversible axonal loss.5,10 However, there is conflicting evidence about the degree of axonal damage in MOG-ON compared to AQP4-ON.5,10,29 We demonstrate less axonal loss in MOG-ON, which correlates with greater preservation of visual acuity in our cohort. This also corresponds with the trend towards maintenance of mfVEP amplitude, a marker of the number of functioning axons in the optic nerve, in MOG-ON compared to AQP4-ON, suggesting more advanced axonal loss in the latter condition. 30 However, axonal damage can accumulate with relapses of MOG-ON, with greater loss of RNFL thickness seen in recurrent cases, consistent with previous results. 10 This could account for the severe visual impairment reported in some MOG-ON patients despite the typically favourable visual outcomes in this group.2–5,8 The clinical implication of this finding is a risk of sustained visual impairment and accumulation of disability with relapses of MOG-ON, highlighting the importance of early and appropriate immunotherapy in this group to induce remission and prevent sustained visual impairment.
In contrast, the effect of relapses on RNFL thickness in AQP4-ON was suggestive but did not meet significance. This would be consistent with the severe axonal loss seen following the initial episode of ON in this condition, 14 with a lesser degree of additional damage resulting from subsequent relapses. In the MS cohort, there were few cases of recurrent ON affecting the same eye and no significant relationship to RNFL thickness could be demonstrated.
Greater mfVEP conduction delay is seen in MS-ON compared to MOG-ON. This is most likely related to the more extensive demyelination occurring along the length of the visual pathway in MS 31 compared to predominant anterior optic pathway involvement in MOG-ON. 2 In contrast, similar mfVEP latency prolongation is seen in both AQP4-ON and MOG-ON. In AQP4-ON, this conduction delay occurs in the setting of profound axonal loss with reduced mfVEP amplitude and thinning of RNFL and can be attributed to degeneration of the fastest conducting fibres in the optic nerve. We propose that in MOG-ON both mfVEP latency prolongation and a degree of amplitude loss can be accounted for by a combination of optic nerve demyelination and conduction block, which produces functional impairment of nerve fibres without such severe axonal loss as AQP4-ON, reflected in relative preservation of RNFL. Although our results were obtained in the chronic post-ON phase, persistent conduction block has been demonstrated in peripheral nerves in multifocal motor neuropathy, including in association with MOG antibodies, and this remains a feasible contributor to the pathophysiology of MOG-ON.32,33
These findings may reflect underlying pathophysiological differences between these conditions. AQP4-NMOSD is an autoimmune astrocytopathy which produces direct retinal damage through effects on Müller cells, astrocytes and retinal vasculature, resulting in neuronal and astrocyte death associated with inflammatory cell infiltration and complement-mediated myelin loss.34,35 This manifests clinically as profound loss of RNFL thickness and risk of permanent visual loss with each episode.14,34 In contrast, MS and MOGAD cause prominent demyelination resulting in functional impairment of conduction along optic pathways, mediated by distinct pathophysiological mechanisms. MS is complex and pathologically diverse, with neuropathological features including inflammatory demyelination, axonal injury and reactive gliosis that affect the posterior visual pathways as well as the optic nerve. 36 Antibody-mediated damage to myelin sheaths is the presumed mechanism of MOGAD. 37 Anatomically, MOG sits in the outermost layer of the myelin sheath and represents <0.05% of total myelin protein. 37 Damage to myelin in MOGAD may be more focal and less severe than in MS, with greater potential for remyelination and recovery. There is also evidence in animal models that anti-MOG antibody binding causes reversible conformational change and internalisation of MOG protein, disrupting myelin structure, causing retraction of oligodendrocyte processes and altering expression of axonal proteins, including those critical to normal function of the node of Ranvier.35,38,39 While the relevance of these pathological mechanisms in human tissue remains to be confirmed, this could potentially help explain the less profound conduction delay in MOG-ON eyes compared to MS-ON as well as the lack of accumulation of demyelination with repeated attacks of MOG-ON in our cohort. Additionally, reversible conduction block induced by anti-MOG antibodies, also demonstrated in animal models, is likely to play a part in the underlying pathophysiology of this disorder. 40 This could also help account for the lesser degree of demyelination seen in MOG-ON compared to MS-ON as well as the rapid clinical response to, and often notable dependence on, steroids, which is now recognised as a hallmark feature of this condition, with relapses often precipitated by rapid steroid weaning or temporally linked with steroid cessation.2,3
Prolongation of mfVEP latency and RNFL loss in fellow non-ON eyes in MS but not AQP4-NMOSD has been previously observed. 14 This is thought to reflect diffuse and bilateral demyelination in visual pathways in MS, with additional neuronal loss also occurring outside of discrete episodes of ON possibly due to retrograde transneuronal degeneration secondary to optic radiation lesions or primary neurodegeneration in the retina.14,18,41 Our data do not demonstrate mfVEP conduction delay or RNFL loss in non-ON eyes in MOGAD, which suggests this could be a distinct electrophysiological marker for differentiation of this disorder from MS. However due to the frequency of bilateral ON in the MOGAD group, there were few non-ON eyes available for analysis and consequently this finding requires further validation in larger cohorts.
Using a multimodal approach to establish patterns of abnormality associated with different underlying causes of inflammatory ON can facilitate diagnosis and enable institution of appropriate therapy. This is critically important due to the vastly different treatment approaches required for these conditions. Immunomodulatory therapy has proven efficacy in MS, however some agents, such as natalizumab, worsen outcomes in AQP4-NMOSD and MOGAD patients.6–8 In contrast, early immunosuppression and plasma exchange in AQP4-NMOSD reduces relapse rates and improves outcomes, and steroids and intravenous immunoglobulin have significant efficacy in MOGAD.1,9 Given the propensity of MOGAD to relapse and the demonstrated association between poor visual outcomes, retinal axonal loss and number of ON episodes, appropriate immunotherapy is of critical importance. 4 Furthermore, our results would support careful consideration of escalating immunosuppression in MOGAD patients with a second episode of ON and evidence of RNFL atrophy and mfVEP amplitude loss despite relative preservation of visual acuity, because this group is at high risk of significant irreversible visual impairment with further relapses. There is also evolving evidence of subclinical progressive retinal axonal loss in the MOGAD group, further emphasising the need for definitive immunosuppression in this group. 42
The present study has several limitations. The sample size of MOG-ON patients was relatively low. Also as a cross-sectional study, causality cannot be established. Future prospective studies across larger patient cohorts from disease onset with longitudinal follow-up are required.
Overall, our study demonstrates differential patterns of retinal axonal loss and functional optic nerve impairment in a MOGAD cohort compared to patients with AQP4-NMOSD and MS. MOG-ON patients experience less severe axonal loss compared to AQP4-ON, with greater preservation of both RNFL thickness and visual acuity. However, recurrent ON in this relapsing disorder leads to gradual accumulation of axonal loss with repeated episodes of demyelination. The total degree of demyelination is significantly less severe in MOG-ON than MS-ON, which is consistent with the more focal anterior visual pathway demyelination and absence of fellow eye abnormalities seen in this disorder. Identification of the underlying aetiology of inflammatory ON using these parameters in combination with clinical and radiological features will enable accurate diagnosis, commencement of appropriate therapy and improved visual prognostication for these patients.
Supplemental Material
sj-docx-1-mso-10.1177_20552173211063126 - Supplemental material for Structural and functional markers of optic nerve damage in myelin oligodendrocyte glycoprotein antibody-associated optic neuritis
Supplemental material, sj-docx-1-mso-10.1177_20552173211063126 for Structural and functional markers of optic nerve damage in myelin oligodendrocyte glycoprotein antibody-associated optic neuritis by Stephanie Barnes, Yuyi You, Ting Shen, Todd A Hardy, Clare Fraser, Stephen W Reddel, Fabienne Brilot, Sudarshini Ramanathan, Alexandr Klistorner and Con Yiannikas in Multiple Sclerosis Journal – Experimental, Translational and Clinical
Supplemental Material
sj-docx-2-mso-10.1177_20552173211063126 - Supplemental material for Structural and functional markers of optic nerve damage in myelin oligodendrocyte glycoprotein antibody-associated optic neuritis
Supplemental material, sj-docx-2-mso-10.1177_20552173211063126 for Structural and functional markers of optic nerve damage in myelin oligodendrocyte glycoprotein antibody-associated optic neuritis by Stephanie Barnes, Yuyi You, Ting Shen, Todd A Hardy, Clare Fraser, Stephen W Reddel, Fabienne Brilot, Sudarshini Ramanathan, Alexandr Klistorner and Con Yiannikas in Multiple Sclerosis Journal – Experimental, Translational and Clinical
Footnotes
Acknowledgements
The authors would like to thank all the patients who participated in this study. The authors would also like to thank Ms Nonna Saakova for her assistance with performing OCT and mfVEPs.
Declaration of conflicting interests
Dr Stephanie Barnes has accepted travel compensation from Merck and Sanofi Genzyme, and fellowship funding from Allergan. A/Prof Todd Hardy has served on advisory boards for Merck, Biogen, Roche and Bristol Myers Squibb, received personal compensation for speaking engagements from Biogen, Merck, Novartis, Sanofi Genzyme and Teva. He has received research/grant support from Multiple Sclerosis Research Australia and Novartis. He serves as co-editor of Advances in Clinical Neuroscience and Rehabilitation. A/Prof Stephen Reddel reports grants and personal fees from Sanofi Genzyme, personal fees and departmental support from Biogen, CSL, and Baxter; and departmental support from Novartis, outside the subject of the submitted work. A/Prof Fabienne Brilot has received funding from the National Health and Medical Research Council (Australia), Multiple Sclerosis Research Australia, the University of Sydney Research Excellence Initiative grant (Australia). She has received honoraria from Biogen Idec and Merck Serono as an invited speaker. Dr Sudarshini Ramanathan has received research funding from the National Health and Medical Research Council (Australia), the Petre Foundation (Australia), the Brain Foundation (Australia), the Royal Australasian College of Physicians, and the University of Sydney. She serves as a consultant on an advisory board for UCB, and has been an invited speaker for Biogen and Excemed. Prof Con Yiannikas serves on advisory boards for Lundbeck and Abbvie.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study would not have been possible without funding support from the National Health and Medical Research Council, the National Multiple Sclerosis Society and the Ophthalmic Research Institute of Australia (ORIA).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.