
Abstract
Rationale:
Due to next-generation sequencing, variants in new genes such as DNAJB11 are recently being identified as causing atypical autosomal dominant polycystic kidney disease (ADPKD). It is important to describe phenotypes associated with these variants in order to increase awareness among clinicians, especially since genetic variability affects ADPKD severity.
Presenting Concerns of the Patient:
We describe a 55-year-old female patient of Haitian origin who presented with slowly deteriorating kidney function, microscopic hematuria, proteinuria, enlarged kidneys with innumerable small cysts, and a family history of chronic kidney disease and cysts. The phenotype was atypical for ADPKD caused by PKD1 or PKD2 variants, since cysts were of small size, kidneys were only moderately enlarged, and the patient had no extra-renal involvement suggestive of typical ADPKD such as liver cysts, pancreatic cysts, cranial aneurysms, or cardiac abnormalities.
Diagnoses:
A panel of genes was analyzed by next-generation massive sequencing techniques, including DNAJB11, DZIP1L, GANAB, HNF1B, PKD1, PKD2, and PKHD1. Genetic testing revealed a heterozygous variant in the DNAJB11 gene (c.123 dup), which is predicted to result in premature protein termination (p. Lys42*) and was classified by the laboratory as likely pathogenic.
Interventions:
She was treated with candesartan 16 mg once daily to address her proteinuria.
Outcomes:
At the time of the most recent follow-up, her proteinuria has increased, and her kidney function continues to slowly deteriorate.
Teaching Points:
DNAJB11 variants are a rare cause of atypical ADPKD. It is important to recognize the clinical features that help distinguish DNAJB11 from PKD1 and PKD2 variants. Atypical ADPKD due to DNAJB11 variants is usually characterized by small cysts, normal kidney size, proteinuria, progressive chronic kidney disease, and phenotypic overlap with autosomal dominant tubulointerstitial kidney disease (ADTKD). It may, however, present itself with enlarged kidneys as was seen in our patient. Genetic testing should be offered whenever a patient presents atypical features of ADPKD, which also requires increased awareness among clinicians regarding the various phenotypes of atypical ADPKD.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a monogenic disease characterized by the development and growth of kidney cysts along with the expansion of kidney volume. 1 With time, this leads to kidney failure (KF), making ADPKD the fourth leading cause of KF worldwide. 2 Most cases of ADPKD are due to variants in 2 genes, PKD1 and PKD2, which encode membrane proteins polycystin-1 (PC1) and polycystin-2 (PC2), respectively. 1 Pathogenic variants to PKD1 and PKD2 are identified in about 72% to 75% and 15% to 18% of genetically positive ADPKD cases, respectively, 2 and cause typical ADPKD, characterized by enlarged kidneys with innumerable cysts, progression to KF, as well as extra-renal features.3-5 In around 10% of cases, no variants in PKD1 or PKD2 are detected. 1 With next-generation sequencing (NGS), new genes are being identified as causing ADPKD, such as GANAB, ALG5, ALG8, ALG9, IFT140, and DNAJB11.2,6-10 Variants in these genes often cause atypical ADPKD, a heterogenous group with usually less severe presentation, as can be seen outlined in Table 1.
Clinical Features of ADPKD Depending on the Affected Gene.

Note. ADPKD = autosomal dominant polycystic kidney disease; ADTKD = autosomal dominant tubulointerstitial kidney disease; KF = kidney failure.
The gene product of DNAJB11 is a glycoprotein cofactor of binding immunoglobulin protein (BiP), which functions as a chaperone in the endoplasmic reticulum for control of folding, trafficking, and degradation of membrane proteins, including PC1 and PC2. 13 Variants in DNAJB11 reduce the functional PC1 dosage within individual tubular epithelial cells and cause cyst formation. 1
Heterozygous pathogenic variants in DNAJB11 have recently been described as causing atypical ADPKD (Table 1).2,11,12 In fact, DNAJB11 variants have been identified in up to 2.6% of the PKD1/PKD2-negative pedigrees of 1 cohort. 12 ADPKD due to DNAJB11 variants is characterized by small renal cysts, resulting in nonenlarged kidneys.2,11,12 Evolution to KF is usually seen after the sixth decade. 2 Also, there seems to be phenotypic overlap between ADPKD and autosomal dominant tubulointerstitial kidney disease (ADTKD) in patients with DNAJB11 variants.2,12 In fact, interstitial fibrosis occurs in noncystic parenchyma, and recurring episodes of gout have been reported.
Liver cysts may develop in DNAJB11-related disease, but no severe liver phenotype has been reported. 13 Vascular phenotypes, including intracranial aneurysms, dilatation of thoracic aorta, and dissection of carotid artery, were noted in 5 patients among 77 cases described by Huynh et al. 12 Abdominal wall hernias were also present in 4 cases. 12 In a retrospective cohort study of 27 patients recently published by Pisani et al, 11 diabetes mellitus was more prevalent compared to typical ADPKD (19% vs 0%; P = .007), whereas cardiac valvular defects were rarer (4% vs 51%; P < .001). Pancreatic cysts were reported in 1 patient, nephrolithiasis was present in 61.5% of patients, and proteinuria, predominantly albuminuria, was found in various degrees, from mild to nephrotic range. 11
Here, we describe a patient who presented with atypical ADPKD due to a pathogenic variant in DNAJB11, with a few different characteristics compared to what has been described regarding ADPKD due to DNAJB11 variants.
Presenting Concerns
A 55-year-old female patient of Haitian origin was seen in our nephrology clinic in 2019 for kidney cysts and microscopic hematuria.
Clinical Findings
The patient had a past medical history of obesity and type 2 diabetes mellitus, which was diagnosed 9 years prior, treated with oral antidiabetic medication and caused mild retinopathy. She had high blood pressure and dyslipidemia, diagnosed 11 years prior. Other medical conditions included uterine fibromas, an umbilical hernia, and a previous cholecystectomy. She had a total of 4 pregnancies, including 1 abortion. Sleep apnea was diagnosed on follow-up, as well as colonic polyps.
As for family history, her mother had high blood pressure and kidney cysts. The patient had a sister who had KF at the age of 55 years, attributed to diabetic nephropathy, in whom kidney cysts were also described. The rest of her family history was unremarkable; her 3 kids were healthy, and there was no notion of liver or pancreatic cysts nor cranial aneurysms. Of note, she was born of a nonconsanguineous union.
Diagnostic Focus and Assessment
At the time of initial evaluation in 2019, her serum creatinine level was 82 mmol/L, with an estimated glomerular filtration rate (eGFR) of 69 mL/min/1.73 m2. Urinalysis showed microscopic hematuria. She also had albuminuria, with an albumin to creatinine ratio of 0.099 g/mmol. A urinary collection was performed, detecting a proteinuria of 0.89 g per day. eGFR tended to decrease moderately over time, with an average decline of 5 mL/min/1.73m2 per year (Figure 1). Proteinuria remained stable at around 0.1 g/mmol until October 2019 and then started to increase, reaching 0.34 g/mmol in June 2022, despite treatment with candesartan. Complementary laboratory tests showed previous hepatitis B infection and a monoclonal gammopathy of unknown significance (IgG kappa at 0.45 g/L, normal serum light chains). Electrolytes, including potassium and magnesium, were normal. Investigations for autoantibodies, including ANA and ANCA, were negative. She had urinary cytology, which came back normal, as well as a cystoscopy, which only revealed cystitis cystica.
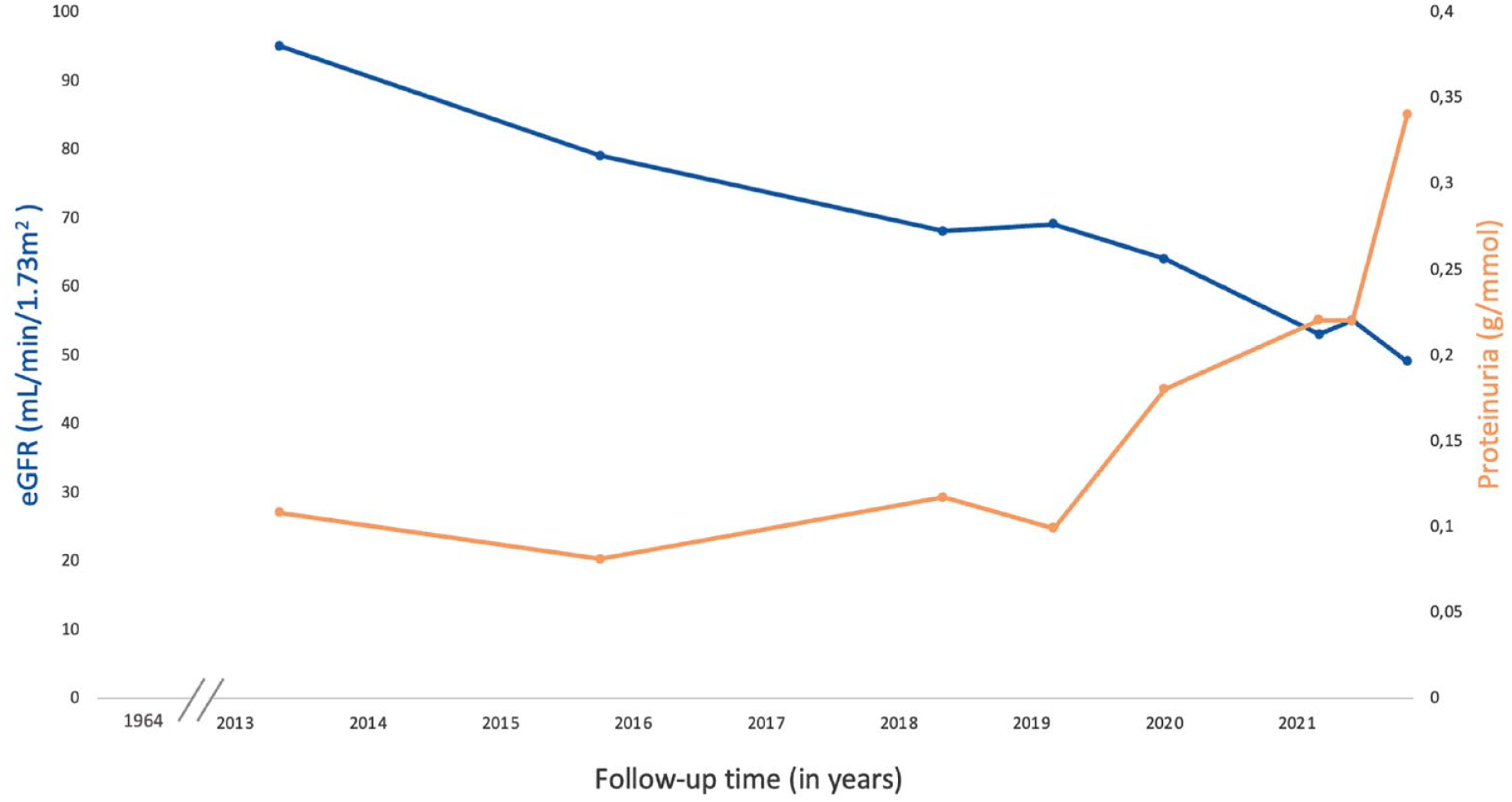
eGFR and proteinuria.
Abdominal ultrasound showed enlarged kidneys (13 cm bilaterally) with multiple cortical infracentimetric cysts, including a few calcified cysts. No liver cysts were detected. Notably, left hydronephrosis was also discovered, for which a mercapto-acetyl-triglycine (MAG3) furosemide renal scan was performed, consisting of simultaneous injection of MAG3, a radiopharmaceutical agent, and furosemide to evaluate the effect of the diuretic on drainage and assess urinary obstruction. 15 The scan showed no significant obstruction.
A computed tomography scan was performed as well, and it showed kidneys of similar size as previous imaging studies with many bilateral cysts, the biggest ones measuring 1.5 cm (Figure 2). The rest of the organ findings were unremarkable, with notably the absence of gynecological or genital tract anomalies aside from uterine fibromas.

Computed tomographic imaging of the kidneys in axial (A) and coronal (B) planes, revealing bilateral small kidney cysts (arrows).
We thus had a 55-year-old patient presenting with slowly deteriorating kidney function, hematuria, proteinuria, innumerable small cysts with modest kidney enlargement, and a family history of chronic kidney disease and cysts. The phenotype was atypical for PKD1 or PKD2, since cysts were of small size, kidneys were only moderately enlarged, and the patient had no extra-renal involvement alluding to ADPKD such as liver cysts, pancreatic cysts, cranial aneurysms, or cardiac abnormalities.3-5
Diabetic nephropathy could explain proteinuria and enlarged kidneys to a certain extent, but hematuria and cysts suggested an alternative diagnosis. In fact, hematuria in diabetic patients usually suggests nondiabetic renal disease. 16 Acquired cysts may be seen in up to 18% of diabetic patients between the ages of 50 and 59 years, but only 28% of these patients have multiple bilateral cysts. 17 A monoclonal gammopathy of renal significance could cause proteinuria and decreased eGFR, although it would not account for the whole phenotype and family history. Given the presence of numerous cysts and slow progression of the nephropathy, renal biopsy was not performed.
Further investigations were carried out to identify a variant explaining the phenotype. Genetic testing was performed at Prevention Genetics, a Clinical Laboratory Improvement Amendments-certified laboratory. DNA was extracted from whole blood specimen. A panel of genes was analyzed by NGS and Sanger sequencing technologies, looking for point variants as well as deletions, duplications, and copy number variants. The following genes were analyzed with a commercial test, chosen according to ClinGen evidence: DNAJB11, DZIP1L, GANAB, HNF1B, PKD1, PKD2, and PKHD1.
Atypical polycystic kidney disease, hydronephrosis, diabetes, and family history made us initially consider a variant in the HNF1B gene. HNF1B-related disease is also inherited in an autosomal dominant manner. It is characterized by renal cysts, along with pancreatic hypoplasia, maturity-onset diabetes of the young (MODY type 5), hyperuricemia and gout, abnormal liver function, hypomagnesemia, and genital tract malformations (Table 1). 14
GANAB variants can cause both ADPKD and autosomal dominant polycystic liver disease, although the predominant phenotype is liver disease, which was absent in our patient. DZIP1L gene impairment is associated with ciliary trafficking defects and renal cystogenesis, causing autosomal recessive polycystic kidney disease (ARPKD). 18 As for PKHD1, it is the primary causative gene for ARPKD.
ALG5, ALG8, ALG9, and recently, IFT140 have also been identified as novel ADPKD genes (Table 1) but were not routinely included in the panel used in our patient at the time of investigation.
Genetic testing revealed our patient to be heterozygous in DNAJB11 gene for a variant (c.123 dup), predicted to result in premature protein termination (p.Lys42*). This variant was classified as likely pathogenic per American College of Medical Genetics and Genomics (ACMG) guidelines. 19
Therapeutic Focus and Assessment
The patient was treated with candesartan 16 mg once daily to address proteinuria and high blood pressure (148/86), as well as amlodipine 10 mg and hydrochlorothiazide 12.5 mg once daily. Since there was little increase in renal volume and the disease was related to a variant in DNAJB11, she was not a candidate for tolvaptan treatment.
Follow-Up and Outcomes
At the time of the most recent follow-up, proteinuria has increased (protein to creatinine ratio 0.34 g/mmol), and her kidney function continues to slowly deteriorate, with an eGFR of 49 mL/min/1.73 m2 (Figure 1).
Discussion
We herein present a case of atypical ADPKD due to a nonpreviously reported pathogenic variant in the DNAJB11 gene, with a few different characteristics compared to what has been noted in the literature.
DNAJB11 variants have been described since 2018 as causing atypical ADPKD, characterized by small renal cysts and resulting in nonenlarged kidneys.2,11,12 These cysts may not be detected by ultrasound but will be revealed by computed tomography or magnetic resonance scans. 11 Some individuals may only have few renal cysts at an advanced stage of nephropathy. 12 This has clinical implications, especially when evaluating a potential living-related kidney donor. Genetic diagnosis seems essential to rule out disease due to DNAJB11 in at-risk patients, and imaging-based diagnostic criteria developed for typical ADPKD should not be used. 12 Interestingly, contrary to ADPKD due to PKD1 and PKD2 variants, normal total kidney volume is not necessarily associated with a good prognosis.2,12 Our patient had in fact small kidney cysts, as seen with this atypical form of ADPKD. However, her kidneys were slightly enlarged. Out of the cases described, 12 other patients had enlarged kidneys as well, with kidney length ≥ 13 cm (up to 19.6 cm in 1 patient).2,12 Although a rare feature of DNAJB11-related disease, enlarged kidneys may be present and do not rule out the disease.
Duplications in DNAJB11, as seen in our patient, have been only exceptionally reported.2,11-13 The duplication carried by our patient had not been previously documented on gnomAD or ClinVar.
There seems to be a phenotypic overlap between ADPKD and ADTKD in patients with DNAJB11 variants.2,12 Analysis of kidney tissue samples from DNAJB11-affected patients showed intracellular retention of UMOD and MUC1 in thick ascending loop epithelial cells. 2 Also, maturation and localization of PC1 were found to be impaired in DNAJB11-null cells. In all, these results suggest that DNAJB11 variants may cause kidney disease through maturation and trafficking defects involving both PC1 and ADTKD proteins, such as UMOD and MUC1. 2 Our patient did not have any gout episodes, which could have helped us suspect a variant in DNAJB11. Since she did not have a kidney biopsy, we do not know whether she had interstitial fibrosis in noncystic parenchyma.
In addition to kidney disease, liver cysts may develop in ADPKD due to DNAJB11 variants, 13 but were not observed in our patient. Abdominal wall hernias were also described, possibly due to altered matrix integrity caused by reduced mature PC1. 12 Of note, our patient had an umbilical hernia. In 1 cohort, diabetes mellitus seemed to be more prevalent in patients with DNAJB11-related disease compared to typical ADPKD. 11 Whether this is merely an incidental finding remains unknown, but it could potentially explain the family history of diabetes mellitus in our patient. However, the diabetes also occurred in the context of her obesity.
Kidney disease severity usually correlates with the variant class and gene involved.1,2 Protein-truncating PKD1 variants are associated with the most severe disease, followed by nontruncating PKD1 variants and PKD2 variants (Table 1).1,2 Progression to KF has not been observed with GANAB variants and seems rare with newly identified IFT140, ALG8, and ALG9 variants, whereas it is seen after the sixth decade with ALG5 variants (Table 1).2,6-10,20 Evolution to KF is usually also seen after the sixth decade with DNAJB11 variants. 2 Our patient is now 58 years old and has mild chronic kidney disease.
Detecting the causative variant in a patient with ADPKD can thus be interesting in several ways. It provides prognostic information, informs the patient about their disease, and is useful for genetic counseling. An accurate diagnosis is also relevant to avoid further investigations, to evaluate a potential family donor for renal transplantation, and, in some cases, to refer to a targeted treatment such as in patients with PKD1 or even PKD2 variants.
Lastly, it is to be noted that ADPKD is currently described as typical or atypical based on our knowledge of different phenotypes. With recent advances in molecular biology, we now know that atypical forms of ADPKD are often due to variants in genes that were not tested in the past and, despite clinical similarities, can vastly differ from typical ADPKD regarding pathophysiology and prognosis. Modifying the classification and designation of these conditions should thus be considered. In that sense, some authors have recently proposed to call this group of disorders the ADPKD-spectrum and to use dyadic terminologies comprising both the clinical condition and gene name when describing the disease (for instance, employing the term ADPKD-DNAJB11 for ADPKD due to DNAJB11 variants).7,9 Other authors have also suggested the use of the term DNAJB11 nephropathy rather than ADPKD, considering the variability in clinical presentation, with some patients having few renal cysts despite advanced kidney disease. 12
In conclusion, our case highlights that DNAJB11 variants should always be included in the differential diagnosis of atypical ADPKD, especially in the context of normal-sized or slightly enlarged kidneys, smart cortical cysts, and slowly deteriorating kidney function. DNAJB11 variants are a rare cause of atypical ADPKD, but prevalence may be underestimated, notably because NGS is relatively new. It is important to recognize the clinical features that help distinguish DNAJB11 from PKD1 and PKD2 variants and acknowledge that patients with variants in the same gene, such as DNAJB11, can present phenotypic heterogeneity.
With the help of NGS, we are now able to identify new variants that explain different phenotypes associated with ADPKD and guide us when counseling our patients. The use of NGS will also allow us to better determine the prevalence of certain variants in the future and possibly help recognize patients with more than 1 variant.
Better characterization of DNAJB11-associated disease may as well be useful to improve our understanding of mechanisms underlying other forms of kidney disease, considering the suspected interplay between DNAJB11 on the one hand and ADPKD and ADTKD proteins on the other hand. Identification of these mechanisms and enhanced comprehension of the pathophysiology of those diseases may hopefully lead to the future development of specific therapeutic strategies.
Footnotes
Acknowledgements
We thank our patient for allowing us to share this case.
Ethics Approval and Consent to Participate
Written informed consent was obtained from the patient for the publication of this case report, in accordance with the requirements of our institution’s ethics committee.
Consent for Publication
Written informed consent was obtained from the patient for the publication of this case report, in accordance with the requirements of our institution’s ethics committee.
Availability of Data and Materials
The data used for this case report are available from the corresponding author upon reasonable request.
Author Contributions
GB and ZE-H were involved in the care of the patient. JK and GB wrote the main manuscript text. All authors contributed to the revision of the manuscript. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.