
Abstract
Purpose of review:
Genetic testing can improve diagnostic precision in some patients with end-stage renal disease (ESRD) providing the potential for targeted therapy and improved patient outcomes. We sought to describe the genetic architecture of ESRD and Canadian data sources available for further genetic investigation into ESRD.
Sources of information:
We performed PubMed searches of English, peer-reviewed articles using keywords “chronic kidney disease,” “ESRD,” “genetics,” “sequencing,” and “administrative databases,” and searched for nephrology-related Mendelian diseases on the Online Mendelian Inheritance in Man database.
Methods:
In this narrative review, we discuss our evolving understanding of the genetic architecture of kidney disease and ESRD, the risks and benefits of using genetic data to help diagnose and manage patients with ESRD, existing public Canadian biobanks and databases, and a vision for future genetic studies of ESRD in Canada.
Key findings:
ESRD has a polygenic architecture including rare Mendelian mutations and common small effect genetic polymorphism contributors. Genetic testing will improve diagnostic accuracy and contribute to a precision medicine approach in nephrology. However, the risk and benefits of genetic testing needs to be considered from an individual and societal perspective, and further research is required. Merging existing health data, linking biobanks and administrative databases, and forming Canadian collaborations hold great potential for genetic research into ESRD. Large sample sizes are necessary to perform the suitably powered investigations required to bring this vision to reality.
Limitations:
This is a narrative review of the literature discussing future directions and opportunities. It reflects the views and academic biases of the authors.
Implications:
National collaborations will be required to obtain sample sizes required for impactful, robust research. Merging established datasets may be one approach to obtain adequate samples. Patient education and engagement will improve the value of knowledge gained.
Why is this review important?
International efforts to develop large biobanks of the general population are underway. Canada’s ethnic diversity and unique populations are opportunities for furthering our understanding of genetics of kidney disease. Patients, clinicians, and researchers will need to work together to translate genetic research into advances in clinical practice.
What are the key messages?
Generation of large collaborative biobanks is required to advance our understanding of the genetic underpinnings of chronic and end-stage kidney disease. Where possible, merging of existing administrative and clinical records is one mechanism to utilize existing data. Opportunity exists for Canada to unite efforts, leading to new discoveries in the genetic basis of kidney disease.
Introduction
Chronic kidney disease (CKD) is a persistent structural or functional abnormality of the kidneys. Over 2.9 million Canadians have CKD, and more than 35 000 have progressed to end-stage renal disease (ESRD). 1 Our ability to successfully identify and treat patients with CKD with the greatest risk of progression to ESRD remains largely limited. ESRD is a heterogeneous outcome resulting from numerous potentially overlapping etiologies and pathophysiologic disease processes, such as hypertension, diabetic nephropathy, interstitial fibrosis, glomerulonephritis, or congenital anomalies of the urinary tract. The cause of ESRD is unknown in 11% of Canadian patients, 2 while many more patients receive a clinical diagnosis without a definitive test (Figure 1). A precision medicine approach that includes traditional history, signs and symptoms, renal biopsy pathology, and biochemical biomarkers, as well as imaging and genetic investigations will contribute to our understanding of how and/or why a patient developed ESRD (Figure 2).3,4 Appropriate investigations in accordance with patient values and economic realities must be considered. Precision medicine may improve diagnostic clarity, prognostic accuracy, and therapy selection and provide valuable information for family planning. In this narrative review, we discuss the evolution of genetic investigations for patients with ESRD and the benefits and challenges of obtaining a genetic diagnosis. We highlight the unique opportunities available to study ESRD genomics through leverage of high-quality health data available in diverse Canadian patient populations.

Stacked bar chart of prevalent ESRD cases in Canada (excluding Quebec) according to Canadian Institute for Health Information data (www.cihi.ca).
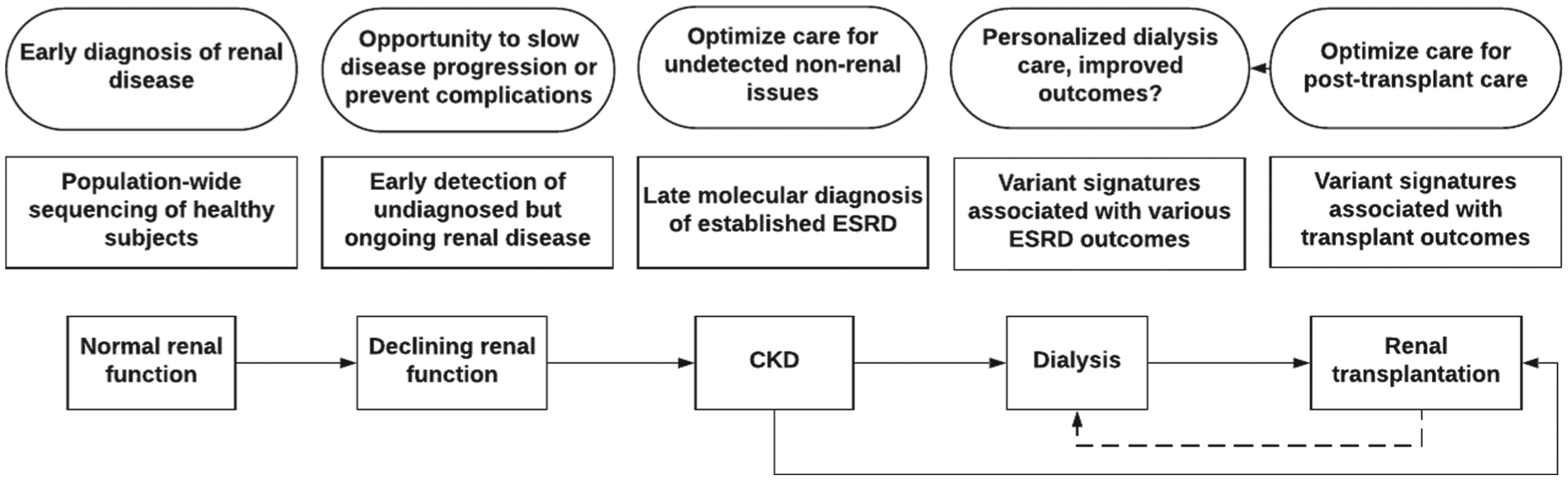
Illustration of the various steps where in-depth genetic analysis may help improve outcomes during the odyssey of a typical patient diagnosed with adult-onset ESRD.
Methods
We sought to review current evidence for the architecture of chronic and end-stage kidney disease and the existing administrative and biobank resources available in Canada. We utilized PubMed searches of English, peer-reviewed articles using keywords “chronic kidney disease,” “ESRD,” “genetics,” “sequencing,” and “administrative databases” and searched for nephrology-related Mendelian diseases on the Online Mendelian Inheritance in Man database. Data tables were generated and information was synthesized into a narrative review. Internal and external peer review was performed as part of the KRESCENT training program.
Review
The Changing Landscape of Genetic Investigations
Advances in technology over the last decade have revolutionized the study of genetics in kidney diseases. Microarray technology allows rapid genotyping of hundreds of thousands of preselected common single nucleotide polymorphisms (SNPs) and was critical to the emergence of genome-wide association studies (GWAS). 5 The development of high-throughput next-generation sequencing in turn enabled rapid genotyping of rare variants. Next-generation sequencing can now read gigabases (×109 base pairs) of sequence in a few hours for a few thousand dollars. Selection of the gene-containing region of the genome (exome sequencing) 6 or targeted genes of interest (gene panels) improves efficiency and reduces cost. 7 Molecular barcoding by ligating a short stretch of manufactured bases to each DNA sample allows for multiplexing many samples into one instrument run, further reducing cost and improving efficiency.
Evolving understanding of genetic kidney diseases
Large population-based GWAS identified thousands of associations between SNPs and quantitative phenotypes or diseases following a “common variant-common disease model” (Table 1). 8 The largest genetic study in nephrology recruited >175 000 participants for a GWAS focused on serum creatinine and estimated glomerular filtration rate (eGFR). This study identified >60 strongly associated loci, yet they only explain a modest proportion of variability in the population (~2%-5%).9,10 These alleles are expected to each have small effect sizes because they are frequent in the general population (minor allele frequencies >5%) and have persisted despite natural selection. It is unclear how these results translate into concrete risk estimation for the development of ESRD because the majority of study participants had eGFR >60 mL/min/1.73 m2. Thus, genotyping these common SNPs currently provides minimal clinically relevant information to individual patients.
Genetic Models of Complex Diseases Including Chronic Kidney Disease and End-Stage Renal Disease.
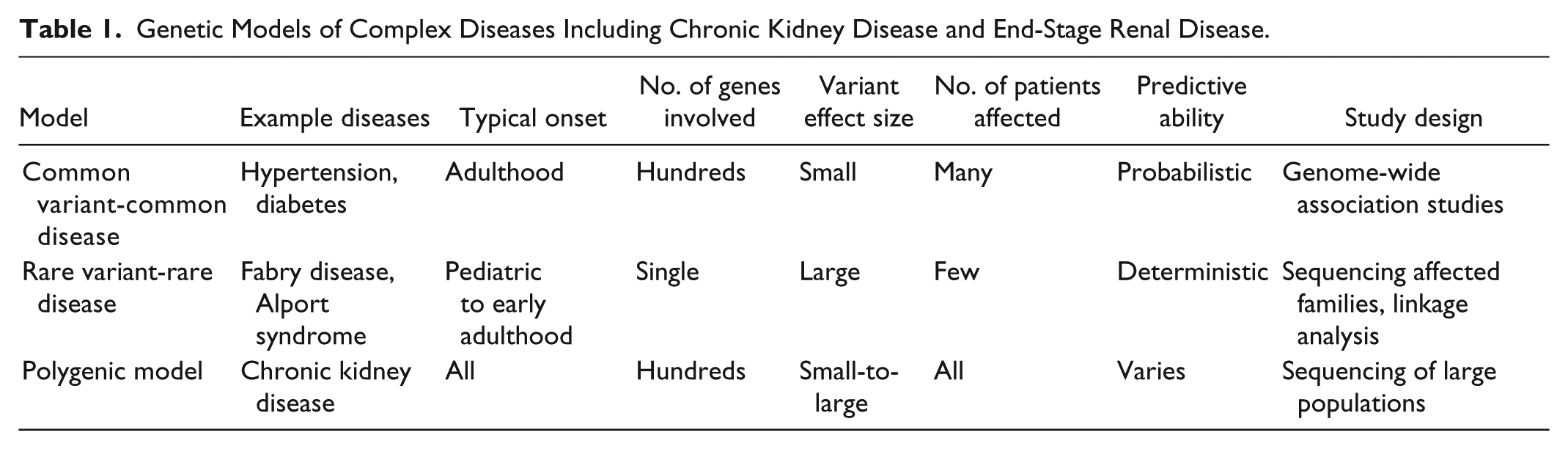
In contrast, the “Rare Disease-Rare Variant model” describes classical Mendelian traits, where a large-effect mutation causes disease with high penetrance following an autosomal dominant, autosomal recessive, or X-linked inheritance pattern. Based on current estimates, 20% of CKD occurring in patients below the age of 25 years are due to a rare mutation from a growing list of genes (Table 2). 11 Both locus heterogeneity (mutations in different genes produce the same disease) and allelic heterogeneity (different mutations in the same gene produce different phenotypes) are common. Generally, population-based GWAS have identified common SNPs with small effect sizes in genes that harbor mutations in Mendelian forms of disease (for example in height, body mass index, diabetes, or lipid levels). 12 However, efforts to identify common variants affecting eGFR in genes that cause rare monogenic renal diseases have been largely unsuccessful, with a few exceptions (UMOD, LRP2, SLC7A9).9,10,13
Growing List of Monogenic Diseases in Nephrology.

Note. Mutations in genes indicated in bold script necessitate reporting as a secondary finding in clinical exome sequencing according to the American College of Medical Genetics and Genomics (ACMG). 14
These two models cumulatively explain a portion of eGFR variability in the general population, and ESRD risk. However, this remains an oversimplification and a combined “polygenic model” is arising as the best explanation. 12 A combination of both protective and deleterious common and rare variants are likely to contribute to the phenotypic expression of even classically defined Mendelian diseases. Digenic compound heterozygosity of deleterious alleles has been observed in polycystic kidney disease 15 and Alport syndrome. 16 Additional mechanisms may also contribute to population trait variability and penetrance, including somatic mosaicism, small noncoding or microRNA, epigenetic regulation, posttranslational modifications, variable X-inactivation, and gene-gene and gene-environment interactions. Finally, it is important to note that individual risk prediction is not directly tied to the proportion of explained variation in the population. For example, a mutation with a large effect on its carrier is likely rare enough to have minimal impact on population trait variability. 12 A deeper understanding of the genetic basis of ESRD will require genetic studies to incorporate a polygenic model, requiring a large amount of data from a representative study population. Large samples will be possible by linking health care administrative databases and biobanks that are developing or in place across Canada.
Genetic Testing in ESRD
While the list of Mendelian kidney diseases has grown significantly in recent years (Table 2), utilization of clinical genetic testing remains low among adult nephrologists. This may be due to 2 common misconceptions. The first misconception is that adult-onset ESRD is almost never due to a genetic mutation. The second misconception is that even if a mutation is discovered, it would be unlikely to alter patient management or disease outcome. Although the prevalence of each Mendelian disease is individually rare, the cumulative population prevalence of all Mendelian kidney diseases is surprisingly high. 17 Strategies for prioritizing ESRD patients for genetic investigations include targeting those with early-onset disease (<45 years old), unclear or absent biopsy results, family history of any renal disease, or presence of extrarenal manifestations. After screening for genetic kidney disease risk factors, a multicenter study applied whole-exome sequencing to ~25% of more than 350 incident patients with CKD, with bona fide pathogenic mutations identified in 22 of 92 (24%) of these patients. 17 Discovery of a genetic etiology of disease can have important ramifications, especially when considering disease recurrence risk posttransplantation. The purpose, consent process, sources of funding, and laboratory standards are different between clinical- and research-based genetic testing. However, they are not completely disparate entities, especially in the context of high throughput gene panel and exome genetic testing. If proper consent is obtained, DNA and genetic information collected for clinical genetic tests can form a rich resource for further genetic research. Conversely, samples collected for genetic research can lead to findings that could have clinical implications in a single participant. Ensuring the consent process is transparent and includes infrastructure and protocols for returning information to patients and research participants is of the utmost importance. Research-based genetic findings should be replicated in a Clinical Laboratory Improvement Amendments (CLIA)–certified laboratory to ensure veracity.
Risks and benefits of identification of a genetic cause of ESRD
Ideally, genetic testing would uncover the cause of CKD early in the course of the disease long before the development of ESRD. Genetic testing may uncover an underlying pathological mechanism, prevent unnecessary investigations including renal biopsy, shorten the diagnostic odyssey, improve risk prediction and stratification, and provide an opportunity for precision therapy. Identification of a mutation may also allow for identification and treatment of sometimes subtle extrarenal manifestations that may have been overlooked. 11 Identification of a genetic cause of ESRD would also allow for low-cost cascade testing of family members, and opportunity for genetic counseling for family planning. It may also help estimate the risk of kidney disease recurrence after renal transplantation. Recognized and unrecognized negative consequences of genetic testing in ESRD also exist. When discussing the benefits of genetic testing with patients, it is important to also highlight some of the potential challenges that it inevitably triggers, such as dealing with incidental findings, risk of genetic discrimination, and interpretation of variants of uncertain significance.
Incidental genetic findings
High-throughput genetic testing, including whole-genome, exome, and gene panel sequencing, has led to issues regarding the reporting of incidental findings. 18 Incidental findings include variants unrelated to the disease under scrutiny, but that could nevertheless be medically relevant to the patient or their extended family. 18 The American College of Medical Genetics recommends a systematic check for pathogenic variants in 59 genes associated with 27 severe but treatable Mendelian conditions when performing clinical exome testing (the subset of the 59 genes relevant to nephrologists are in bold in Table 2). 14 Variants in these genes are now defined as “secondary findings” as they must be actively sought out, 14 and “incidental findings” encompass pathogenic variants found in other genes throughout the genome. In research settings, it is important to clarify if the patient wants to be made aware of secondary or incidental findings as they have the right to opt out.19-22 Secondary findings are common, as whole-genome sequencing data from 1000 healthy adults revealed that ~1% had a mutation in one of these genes.23,24 These issues must be taken into account when planning large-scale sequencing projects to minimize liability and budget for the additional costs triggered by these disclosures (eg, genetic counseling).25,26
Variants of uncertain significance
The American College of Medical Genetics provides guidelines to assess the pathogenic potential of rare missense variants. This exercise remains quite challenging, and many cases that remain inconclusive are labeled “variants of uncertain significance.” Comparison with large sequence databases is the first step, as >99% of causative rare variants have minor allele frequencies below 0.01%. 27 Delineation of the co-segregation pattern of a rare variant with affection status in families can be helpful, but is often impractical or not possible. Bioinformatics tools can estimate a variant’s pathogenic potential, but unfortunately they frequently overestimate the pathogenicity of variants. 28 Functional validation of variants using in vitro or in vivo models remains the gold standard, but it is very costly and time-consuming. At present, the pathogenicity of the vast majority of variants found during genetic testing is unclear.
Genetic discrimination in Canada and abroad
The acceptance and use of genetic testing in clinical and research settings may be dramatically hampered if genetic discrimination is not prevented. Genetic discrimination is defined as “adverse treatment that is based solely on the genotype of asymptomatic individuals.” 29 For example, health insurers have declined to offer coverage for at-risk individuals who disclosed genetic information, including family history. 30 There have also been cases where employers have used this information to dismiss potential or current employees. 31 All members of European Union enacted legislation against genetic discrimination in the 1990s, 32 as did the United States in 2008 with the passage of Genetic Information Nondiscrimination Act (GINA).33,34 Canada remained the only G7 country without clear genetic discrimination legislation until the Genetic Non-Discrimination Act became law in Canada in May 2017.35,36 This had real consequences as Canadians were routinely refused life and disability insurance on the basis genetic risk factors or family history. 37 Similar problems have occurred in other developed nations with national health care systems, such as Japan 38 and Australia. 39 In contrast to European and American legislations, the Canadian Genetic Non-Discrimination Act adopted a narrower definition of “genetic information,” exclusively focusing on genetic testing results. Indeed, clinical information and family history are not protected under the Genetic Non-Discrimination Act even though these data provide information about the genetic makeup of an individual. While the Genetic Non-Discrimination Act is undeniably a positive development toward implementation of more widespread genetic testing in Canada, life and health insurers could use this loophole for lawful genetic discrimination. In sum, patients will need to weigh the potential risks and benefits from participation in genetic studies of ESRD.
Linking Canadian Administrative Databases and Biobanks for Genetic Research
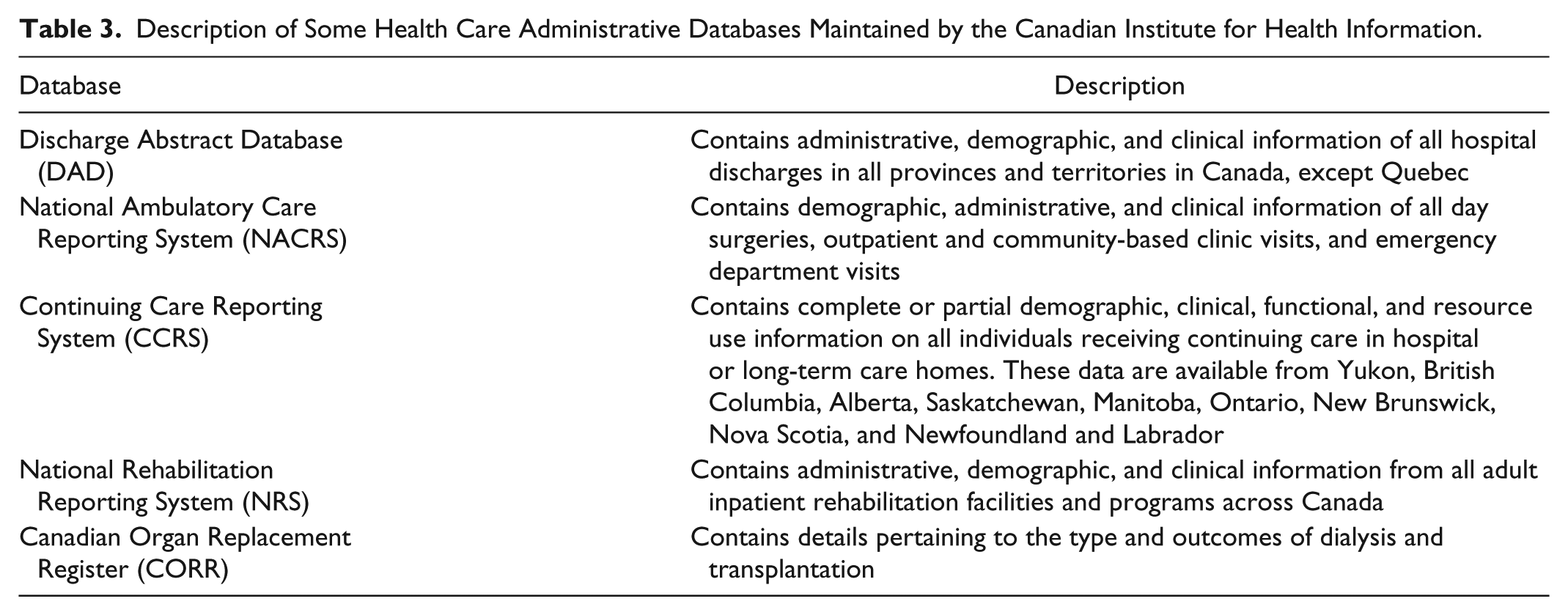
Canada’s universal and publicly funded health care system provides a wealth of administrative data to researchers. Each citizen has a unique health identifier, allowing for reliable data linkage across databases. 40 Administrative data provide inexpensive access to large observational, population-based datasets with long follow-up times. The Canadian Institute for Health Information (CIHI) is a nonprofit organization that collects and analyzes these data and facilitates public access to health data (Table 3). 41
Description of Some Health Care Administrative Databases Maintained by the Canadian Institute for Health Information.
In parallel, investigators from across Canada have developed private biobanks which include genetic testing data. Canada’s largest prospective biobank initiative is the Canadian Partnership for Tomorrow Project (CPTP) that includes subjects from 5 cohorts recruited in 8 provinces (Table 4). 42 The aim of CPTP is to identify environmental, lifestyle, and genetic risk factors of diseases, including CKD. 43 The CPTP plans to continue to collect health information until at least the year 2033 and will ultimately include a myriad of data points for hundreds of thousands of Canadians. 42
Regional Cohorts of the Canadian Partnership for Tomorrow Project (CPTP).

Studies linking biobanks to administrative data can overcome the shortcomings of each type of data source. Existing biobanks generally lack the detailed health care utilization data available in administrative databases, and administrative databases lack biological information found in biobanks. It is important to acknowledge that administrative data are not collected with the same rigor used in research studies; therefore, the quality and reliability of the data can be affected. 44 Researchers who use health care administrative data typically identify diseased patients and build study cohorts using the International Classification of Disease, Ninth Revision (ICD-9) or Tenth Revision (ICD-10) codes, or physician claim diagnosis codes. Although ICD-9 and ICD-10 codes are highly specific, they are not sensitive because they only identify patients with a hospital admission or emergency department visit who usually have a more advanced disease.45,46 In contrast, physician claim diagnosis codes generally have poorer specificity but greater sensitivity to capture a much higher percentage of the relevant population. Linking genetic information from biobanks to administrative databases would allow researchers to assemble study cohorts with definitive diagnoses and broader disease spectra. It would also provide invaluable phenotypic information for large-scale genotype-phenotype association studies.
Opportunities and barriers to merging health data across jurisdictions
Merging health data (including administrative data and biobanks) across provincial and/or territorial jurisdictions could increase research capacity in Canada. For example, merging databases allows for assembly of larger cohorts of patients that may include several individuals with rare conditions. Merging may also help avoid duplication of efforts, such as having multiple investigators collecting the same data to answer overlapping research questions. It could also increase researchers’ access to data from jurisdictions that are outside of their home province/territory. For example, the Ontario Drug Benefits dataset contains prescription drug claims limited to a small subset of the population that primarily consist of individuals over the age of 65 years. In contrast, every Québecers without private prescription drug insurance is covered under the public drug insurance plan (Régie de l’Assurance Maladie du Québec) and thus contains information from patients with a more diverse age range. Broad access to these 2 databases for Canadian researchers (especially if merged) would allow them to ask unique questions using the most relevant data available.
Unfortunately, several barriers prevent merging of provincial and territorial health care administrative data. The major barriers that currently exist and potential solutions to circumvent them are summarized in Table 5.40,47,48 A report published by CIHI in 2002 provides a detailed account of specific legislative barriers to data linkage across Canadian jurisdictions that is still relevant. 48
List of Few Barriers to Link Data Across Jurisdictions in Canada and Potential Approaches to Overcome the Barriers.

There are several initiatives to link biobanks to administrative databases in Canada. For example, the British Columbia (BC) Generation Project is linked to administrative Population Data BC on an ongoing basis. 49 We should continue to populate existing biobanks, to establish new regional cohorts in jurisdictions not currently involved in CPTP, and continue linking biobanks with administrative databases. The success of the continual expansion of biobanks and linkage to administrative databases for genetic research is highly reliant on Canadians’ willingness to share their detailed medical and genetic information. Engaging patients can help with this and with genetic research in general.
Patient Engagement to Facilitate Proliferation of Genetic Research in Canada
Patient engagement occurs when researchers, patients, and caregivers collaborate “in the governance, priority setting, and conduct of research, as well as in summarizing, distributing, sharing, and applying its resulting knowledge” (ie, knowledge translation). 50 Studies show that patient engagement leads to higher enrollment and retention rates and dissemination of findings in a more meaningful and understandable way. 51 The Canadians Seeking Solutions and Innovation to Overcome CKD (Can-SOLVE CKD) network’s experience also emphasizes the value of patient engagement. 52 The Can-SOLVE CKD network is a unique partnership between patients, researchers, practitioners, and policy makers to revolutionize the care of Canadians with CKD and is one of 7 networks funded under the Canadian Institutes for Health Research Strategy for Patient-Oriented Research initiative. Patient engagement lead to identification of top patient-relevant research priorities that created the foundations for the 18 research projects covered under the umbrella of Can-SOLVE CKD. 52
Patient engagement is of particular importance in genetic research as it relies on patients’ willingness to share medical and genetic data. Assembling biobanks and linking them to health care administrative databases requires broad participant consent for use of both biological specimens and data for research. Patient collaborators are often willing to participate in all stages of the research process including setting research priorities, study design, research dissemination, and knowledge translation. They may provide insight into reasons that could deter others from participation and advise on methods to communicate incidental findings and how to communicate the issue with prospective participants during patient recruitment. Patient collaborators can help draft and revise consent forms to make them understandable to future participants and help communicate the importance of future genetic research when recruiting new patients. In addition, patient collaborators can share their experiential knowledge. Finally, they may help advocate to improve the Canadian Genetic Non-Discrimination Act. Patient engagement is essential for the development of robust genetic studies focused on ESRD.
Avenues for Future Genetic Study of ESRD
Enrollment of large samples of patients in biobanks with linked administrative health data will provide the opportunity for study designs that include both common and rare genetic variants for the evaluation of both common quantitative traits and rare Mendelian diseases. Should investigators despair that sample sizes of hundreds of thousands of participants are required for novel genetic discoveries? Using extreme phenotypes, especially unaccounted for by traditional risk factors, is one strategy to increase power. 53 For example, one could compare patients with diabetic nephropathy who develop early ESRD despite excellent risk factor management to those who progress slowly despite the presence of such risk factors. Furthermore, knowledge of a genetic association with phenotypes in the general population can be leveraged in the ESRD population. For example, variants associated with elevated fasting blood sugar or hemoglobin A1C in the general population could be tested for association with risk of developing new onset diabetes after kidney transplantation.
Canada’s ethnic diversity imposes both challenges and opportunities for genetic studies of ESRD. Databases of genetic data from control subjects are heavily focused on European and African American subjects limiting interpretation of rare variants in other populations. However, the multiethnic Canadian landscape provides a unique opportunity for transethnic and admixture association and mapping studies. 54 Efforts to collate patients into extended pedigrees in databases will greatly improve power for studying rare variants in ESRD. Collaboration between centers to facilitate collection of patients into consortia creating the largest possible sample sizes is a necessity.
Research efforts using next-generation sequencing, uncovering Mendelian forms of CKD in ESRD, are currently of great interest. In contrast, do small effect common genetic variants even matter? Interest in GWAS has waned after the proliferation of next-generation sequencing, but many questions amenable to GWAS remain in nephrology. GWAS-derived common variants can provide insight into biology and potential therapeutic targets, but pinpointing the responsible genes in GWAS-identified regions is challenging and ongoing. Identified genes can be used as drug targets for creating larger effects than the observed effect of the SNP in the GWAS. GWAS-derived SNPs can be combined into polygenic risk scores, and patients identified with a rare collection of many common risk variants may present with a phenotype similar to those with Mendelian disease. Mendelian randomization studies jointly test the association between genetic variants and both a risk factor and an outcome to support that the risk factor is a causative contributor to the outcome. For example, reduced high-density lipoprotein (HDL) cholesterol appears a risk factor for CKD, as genetic variants associated with reduced HDL are also associated with CKD risk. 55 However, Mendelian randomization requires GWAS-identified genetic predictors of the risk factor. In sum, incorporating both common small effect and rare large effect variants, as well as other sources of “missing heritability,” will be required to maximize the insight gained from genetic studies in ESRD.
Using genetic variants to optimize renal replacement therapy
The potential contribution of genetics to hemodialysis and peritoneal dialysis (PD) outcomes remains inadequately studied. Evidence is limited to candidate gene association studies, which have significant issues with reproducibility and publication bias. Studies looked for SNP associations with hemodialysis outcomes including vascular access issues,56-61 biochemical indices,62-65 inflammation,66-68 cardiovascular comorbidities66,69-75 and mortality76-79 and PD outcomes including peritoneal transport characteristics,80-84 peritonitis risk,85-87 or risk of encapsulating peritoneal sclerosis 88 (Supplemental Table 1). Sample sizes were inadequate to confidently identify variants of reasonable effect sizes (average sample size of 246, range 67-777 patients). Only a single published GWAS relating to hemodialysis exists, published in 2011, examining survival in 647 African American ESRD patients with type 2 diabetes. 89 No replication study has been published, nor are additional studies reportedly underway. Two ongoing studies could impart knowledge on PD. The first, PD-CRAFT (NCT02042768), is collecting clinical and genetic data on 1495 PD patients. The second, BIO-PD (Biological Determinants of Peritoneal Dialysis; NCT02694068), has an estimated enrollment of 4865 patients between 2014 and 2019. Both studies are part of the Peritoneal Dialysis Outcomes and Practice Patterns Study (PDOPPS), an international consortium formed to promote research aimed at improving practice and outcomes in PD. 90 Genetic studies could improve our knowledge of renal replacement therapy pathophysiology and hold the potential for improving ESRD therapy.
Looking Forward
Genetics could reduce diagnostic ambiguity and contribute to a precision medicine approach to ESRD care. Our understanding of the genetic architecture of complex diseases is growing rapidly, and these advances are applicable to ESRD. Canadians should build a rich data repository for health research, linking and merging health data including biobanks and administrative and clinical databases, across jurisdictions. To facilitate the proliferation of large-scale genetic studies in Canada, it is imperative to overcome the barriers to data linkage by standardizing coding practices, legislation, policies, ethics, privacy and confidentiality review protocols, and genetic testing. Canadians should be protected by broadening genetic nondiscrimination legislation. It is important for patient representatives to be actively involved in setting the priorities for CKD genetic research because they are the ones that stand to benefit the most from this research. Large sample sizes will be required to draw robust conclusions. Artificial intelligence and machine learning algorithms could be applied to such data sources. Systemic changes are needed to ensure equitable attribution of credit to those who build, contribute, analyze, publish, or disseminate knowledge generated from datasets derived from linked databases. Building common biobank and database resources for improved study designs should be embraced by the Canadian nephrology community.
Supplemental Material
Supplemental_table_1 – Supplemental material for Opportunities and Challenges for Genetic Studies of End-Stage Renal Disease in Canada
Supplemental material, Supplemental_table_1 for Opportunities and Challenges for Genetic Studies of End-Stage Renal Disease in Canada by Vinusha Kalatharan, Mathieu Lemaire and Matthew B. Lanktree in Canadian Journal of Kidney Health and Disease
Footnotes
List of Abbreviations
Can-SOLVE CKD, Canadians Seeking Solutions and Innovation to Overcome CKD; CKD, chronic kidney disease; CIHI, Canadian Institute for Health Information; CPTP, Canadian Partnership for Tomorrow Project; eGFR, estimated glomerular filtration rate; ESRD, end-stage renal disease; GINA, Genetic Information Nondiscrimination Act; GWAS, genome-wide association study; HDL, high-density lipoprotein; ICD-9, International Classification of Disease, Ninth Revision; ICD-10, International Classification of Diseases, Tenth Revision; PD, peritoneal dialysis; SNP, single nucleotide polymorphism.
Online web resources
Ethics Approval and Consent to Participate
No patient consent was required for this narrative review.
Consent for Publication
The authors have consented publication of this article.
Availability of Data and Materials
No additional data or materials are available for this review. Please contact corresponding author with any requests.
Authors’ Note
Vinusha Kalatharan, Mathieu Lemaire, and Matthew B. Lanktree contributed equally to conception, drafting, and revision of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: V.K. is supported by a doctoral fellowship award, M.B.L. by a postdoctoral fellowship award, and M.L. a New Investigator award from the KRESCENT Program, a national kidney research training partnership of the Kidney Foundation of Canada, the Canadian Society of Nephrology, and the Canadian Institutes of Health Research. V.K. is also supported by the Canadian Institutes of Health Research Doctoral Scholarship, M.L. is also supported by a biomedical research grant from the Kidney Foundation of Canada, and M.B.L. is also supported by a Ben Lipps postdoctoral fellowship award of the American Society of Nephrology.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.