
Abstract
Rationale:
Alport Syndrome (AS) is a progressive genetic condition characterized by chronic kidney disease (CKD), hearing loss, and eye abnormalities. It is caused by mutations in the genes COL4A3, COL4A4, and COL4A5. Heterozygous mutations in COL4A4 and COL4A3 cause autosomal dominant Alport Syndrome (ADAS), and a spectrum of phenotypes ranging from asymptomatic hematuria to CKD, with variable extra-renal features. In the past, heterozygous mutations in these genes were thought to be benign, however recent studies show that about 30% of patients can progress to CKD, and 15% can progress to end stage kidney disease (ESKD).
Presenting Concerns:
We present a case of a woman who was noted to have microscopic hematuria pre-living kidney donation. Genetic testing revealed a heterozygous variant of uncertain significance (VUS) in the COL4A4 gene. VUSs are medically nonactionable findings and data show that VUSs can be detected in 41% of all patients who undergo clinical genetic testing. VUSs frustrate clinicians and patients alike. Although they cannot be used in medical decision-making, data suggest that reanalysis can result in the reclassification of a VUS over time.
Diagnosis:
Post-donation, the index patient had a higher than anticipated rise in serum creatinine, raising a concern for possible intrinsic kidney disease. Kidney biopsy was deemed high risk in the setting of a unilateral kidney thereby limiting possible diagnostic intervention to determine the cause of disease.
Intervention:
Re-evaluation of prior genetic testing results and reassessment of the previously identified VUS in COL4A4 was performed 5-years post-donation. These analyses, along with the addition of new phenotypic data and extended pedigree data, resulted in the reclassification of the previously identified VUS to a likely pathogenic variant.
Outcomes:
This case demonstrates the importance of structured, periodic re-evaluation of genetic testing results. With the ever-changing landscape of genetics in medicine, the interpretation of a VUS can be dynamic and therefore warrant caution in living kidney donor evaluations. Studies have shown that about 10% of VUSs can be upgraded to a pathogenic classification after an 18- to 36-month interval. Structured re-evaluation of genomic testing results has not yet been integrated into clinical practice and poses a unique challenge in living kidney donation.
Novel findings:
This case report highlights the variability of the ADAS phenotype caused by pathogenic heterozygous variants in the type 4 collagen genes. It supports the nomenclature change from a benign hematuria phenotype to ADAS, particularly when additional risk factors such as proteinuria, focal segmental glomerulosclerosis or glomerular basement membrane changes on kidney biopsy are present, or as in this case, evidence of disease in other family members.
Introduction
Alport Syndrome (AS) is a progressive genetic condition characterized by chronic kidney disease (CKD), hypertension, hearing loss, and eye abnormalities. 1 AS is caused by pathogenic and likely pathogenic mutations in genes that encode the type IV collagen alpha chains: alpha chain 3 (COL4A3), alpha chain 4 (COL4A4) and alpha chain 5 (COL4A5). 1 AS has a X-linked mode of inheritance when a pathogenic mutation occurs in the COL4A5 gene, and autosomal inheritance including dominant, recessive or digenic for COL4A3 and COL4A4 mutations.1,2 Heterozygous mutations in COL4A4 and COL4A3, now known to cause autosomal dominant Alport syndrome (ADAS), result in a spectrum of phenotypes ranging from hematuria to CKD, and to a lesser extent, ocular abnormalities and sensorineural deafness. 3 Originally considered a benign phenotype, 4 it is estimated that 20% of ADAS cases will progress to ESKD when risk factors are present. 3 These include proteinuria, focal segmental glomerulosclerosis (FSGS) or glomerular basement membrane (GBM) irregularities including thinning, thickening, and lamellation on kidney biopsy, a positive family history or other genetic modifiers. 3
A molecular diagnosis can be transformative for patients, leading to personalized treatment plans, better understanding of their disease, a confirmed diagnosis, and accurate genetic counseling. 5 It also allows for cascade testing of family members, family planning options, and it can ultimately ease the burden of uncertainty. 5 In some cases, a genetic diagnosis can help guide decisions during the transplant assessment process for biologically related living kidney donors and recipients alike, especially when there is a positive family history of CKD. 1 With the recent reduction in cost, a genetic diagnosis is available to more patients than ever before, and therefore could be more readily implemented into clinical care. 6
One of the main cited barriers to genetic testing is the concern regarding detection of variants of uncertain significance (VUS). 7 VUS means that there is currently not enough existing data to prove the classification of the variant as pathogenic/likely pathogenic, nor benign/likely benign, following the American College of Medical Genetics (ACMG) guidelines. 8 VUSs frustrate clinicians and patients, as they cannot be used in medical decision-making. 7 Living donation is however a unique situation and to mitigate risk, comprehensive investigation of all potential clinically relevant VUSs is warranted. Unfortunately, due to the time sensitive nature of living donation, we often don’t have the appropriate tools or data at the time to determine the exact significance. 9 Studies show that reanalysis can lead to a 10% to 22% increase in diagnostic rate following integration of new data which include updated pedigree information, improved prediction tools, functional studies, and the discovery of new genes and diseases. 9 - 11 Reanalysis is now recommended at a 1.5 to 3 years interval; however, this timeframe may limit the utility in potential living kidney donors.10,11
Here, we present a living kidney donor case which highlights the importance of periodic genetic re-evaluation and opens the debate on when and how re-evaluation of genetic data should occur in living donors.
Presenting Concerns
The proband (P12) is a 50-year-old Caucasian female who donated her left kidney as a nondirected altruistic donor at the age of 45. Past medical history was significant for altruistic partial liver donation. At the time of donor assessment, her serum creatinine was 66 μmol/L (normal range 53-97.2 µmol/L), estimated glomerular filtration rate (eGFR) 101 mL/min/1.73m2 as measured by the CKD-EPI equation. 12 Persistent microscopic hematuria without proteinuria was noted pre-donation; however, renal imaging and cystoscopy pre-donation were normal. Given her positive family history of CKD in a paternal uncle, genetic testing was performed. This revealed a heterozygous VUS in COL4A4; a guanine to adenine base pair change at position 3307 predicted to result in an amino acid change from glycine to arginine at position 1103 (c.3307G>A, p.G1103R). Since a VUS is not considered a clinically actionable finding, she elected to proceed with donation.
Initial follow-up post donation was largely unremarkable, and she remained normotensive throughout follow-up. Two years post-donation, she developed symptoms suggestive of inflammatory arthritis. Serum creatinine at the time was 115 μmol/L, consistent with an eGFR of 50 mL/min/1.73m2. Radiological imaging of the chest revealed bilateral mediastinal and hilar lymphadenopathy. On review she was noted to have diffuse adenopathy (cervical chain, supraclavicular, hilar, and mediastinal) and was experiencing constitutional symptoms including weight loss, fever, night sweats, low energy, and fatigue. She therefore underwent endobronchial ultrasound-guided transbronchial needle aspiration and pathology. Pathological examination of a subcarinal lymph node demonstrated multiple epithelioid nonnecrotizing granulomas consistent with the diagnosis of sarcoidosis. She was commenced on a tapering dose of prednisone with improvement in both symptomology and radiographic features of adenopathy. She was subsequently maintained on methotrexate therapy. Over this time, she had a progressive rise in creatinine from 115 to 127 μmol/L (eGFR decrease from 50 to 44 mL/min/1.73m2), which prompted a referral to nephrology.
Clinical Findings
Upon referral it was noted that her urine dipstick remained positive for microscopic hematuria but negative for proteinuria, with a urine albumin creatinine ratio of 0.5 mg/mmol (normal range <1 mg/mmol). Serum calcium was normal for the duration of follow-up and 24-hour urine calcium level was normal at 3.65 mmol/day (normal range 2.50-7.50 mmol/day). Ultrasound scan (USS) pre-donation was not available but post-donation USS showed a surgically absent left kidney. The right kidney was normal in size and echogenicity. No hydronephrosis, renal stones, or renal masses were noted. Given the presence of a unilateral kidney and the absence of proteinuria, the joint decision was made not to proceed with kidney biopsy. System review revealed reduced visual acuity with evidence of myopia and astigmatism on ocular assessment. There was no hearing impairment. Family history was updated and revealed hearing loss and reduced visual acuity in the probands father (P7), microscopic hematuria and hearing loss in an adult daughter (P14) and microscopic hematuria in another adult daughter (P16) (Figure 1).
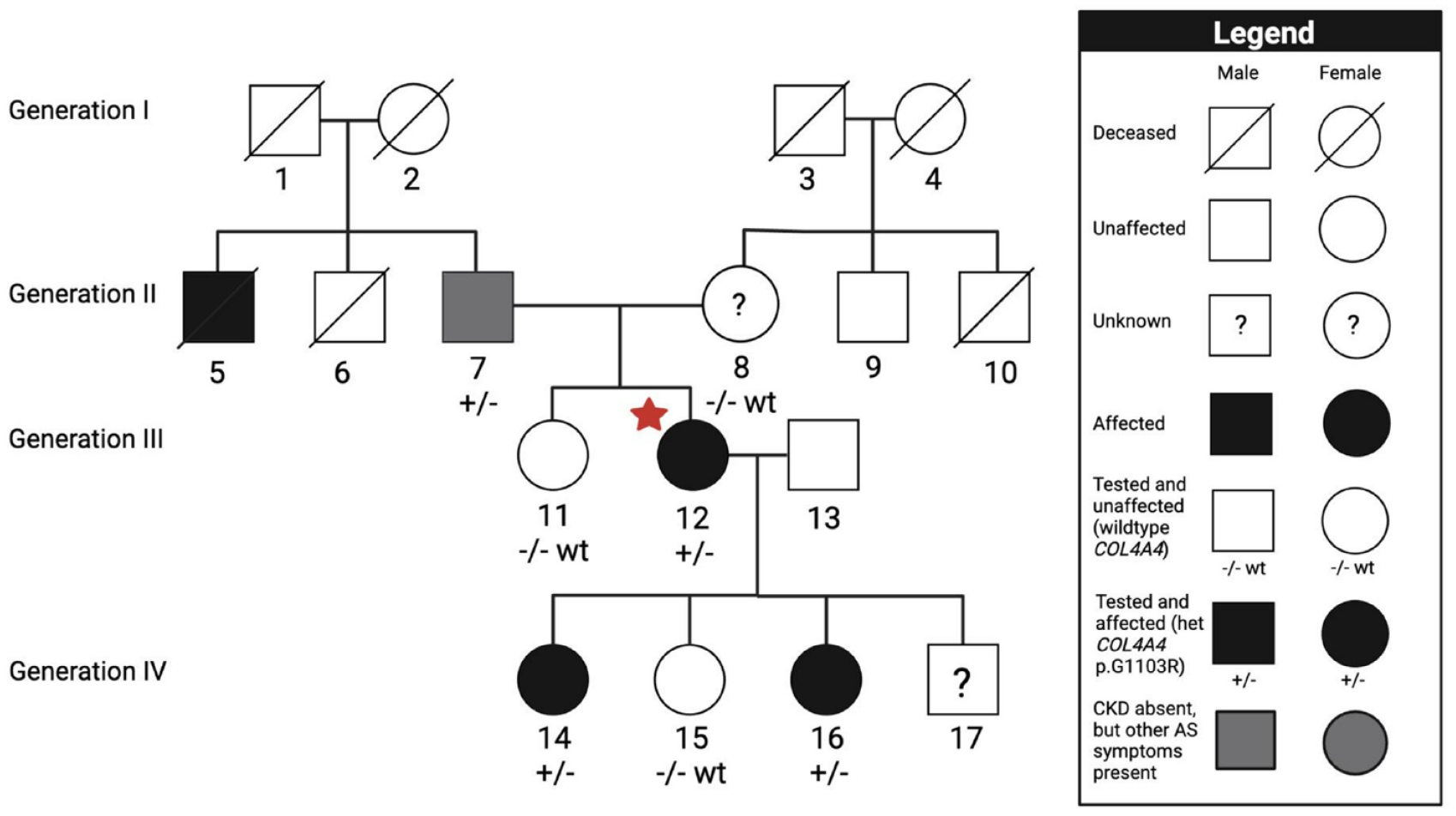
Pedigree analysis and Sanger sequencing results.
Diagnostic Focus and Assessment
Diagnostic Methods
Genetic testing methods
A reanalysis of a previous Alport gene panel was performed in the proband through SickKids Division of Genome Diagnostics, Toronto, Ontario. 13 Briefly, next generation sequencing was performed using Agilent SureSelect capture followed by paired-end sequencing using the lllumina sequencing platform variant calls are generated using Genomic Analysis Tool Kit (GATK) after read alignment with the Burrows-Wheeler Aligner (BWA) to genome build GRCh37/UCSC Hg19 with decoy. Reanalysis was performed in the following 3 genes: COL4A3 [NM_000091.4], COL4A4 [NM_000092.4], and COL4A5 [NM_000495.4]. For at risk family members, specific mutation confirmation using clinical grade testing was performed looking only for the presence or absence of the familial variant in question: COL4A4 c.3307G>A, p.G1103R. This testing was performed either through SickKids Division of Genome Diagnostics or through targeted variant testing by Prevention Genetics, Marshfield, Wisconsin, USA, depending on the location of the family member. For this testing, polymerase chain reaction (PCR) was used to amplify the necessary exons plus additional flanking noncoding sequence. After cleaning of the PCR products, cycle sequencing was carried out using the ABI Big Dye Terminator v.3.1 kit. PCR products are resolved by electrophoresis on an ABI 3730xl capillary sequencer.
Diagnostic Results
Upon re-evaluation of COL4A4, in conjunction with new clinical data and additional data in the literature the COL4A4 c.3307G>A, p.G1103R variant was reclassified from a VUS to likely pathogenic variant. This variant is predicted to cause a glycine substitution in a Gly-X-Y triple helical domain of COL4A4. The glycine substitution in the Gly-X-Y triplet is a common mechanism of disease and is expected to cause disease based on fibrillar collagen biology. 14 The mutation occurs in the 36th exon in a collagen domain (Figure 2). Family testing showed that the probands father (P7), and two adult affected daughters (P14 and P16) carried the same familial variant while the probands unaffected mother (P8), sister (P11), and daughter (P15) were negative.

The variant now meets the pathogenic criteria based on the following ACMG criteria (Table 1):

Note. CADD = Combined Annotation Dependent Depletion; NM = Neutrophil Migration; SIFT = Sorting Intolerant From Tolerant; ACMG = American College of Medical Genetics; SNP = single nucleotide polymorphism; ID = identification.
Adenine (A), American College of Medical Genetics (ACMG), Arginine (Arg), Chromosome change (c. Change), Combined Annotation Dependent Depletion (CADD), deleterious (D), Exome Aggregation Consortium (ExAC), Genome reference consortium human build 37 position (Hg19Pos), Glycine (Gly), Guanine (G), neutrophil migration (NM_), polymorphism phenotyping v2 (Poly2), protein change (p. Change), reference SNP (rs), single nucleotide polymorphism identification (SNP ID), Sorting Intolerant from Tolerant (SIFT), The Genome Aggregation Database (gnomAD)
Computational evidence with prediction tools supporting a deleterious effect of the variant (PP3).
Low allele frequency in population controls (gnomAD) with the variant is absent from controls (PM2).
Reputable sources report the variant as pathogenic, for example, reported as pathogenic in ClinVar (ID 551160) and reported in a homozygous and compound heterozygous state in multiple individuals with Alport syndrome15,16 (PP5).
The patient’s updated phenotype and family history is specific for a diagnosis with a single genetic etiology (PP4).
The prevalence of variant in affected individuals is increased compared to controls with segregation among affected individuals in the family (PS4).
Discussion
We report a case of a woman carrying a heterozygous variant in the gene COL4A4, which was upgraded from a clinical nonactionable VUS pre-donation to a likely pathogenic medically actionable mutation post-donation. Normal physiology post-donation results in an immediate decrease in eGFR and concurrent rise in creatinine, followed by a compensatory increase in the eGFR and concurrent fall in creatinine. 17 In this case, the donor had a greater than anticipated rise in creatinine post-donation, persistent microscopic hematuria in association with new onset myopia and astigmatism as she approached 50 years of age. These findings resulted in the re-evaluation of the COL4A4 variant and subsequent diagnosis of ADAS.
In this case, re-evaluation was serendipitous rather than part of a structured re-evaluation process. However, reclassification of variants can have clinical outcomes for patients and at-risk family members. In this case, confirmation of the diagnosis of ADAS resolved diagnostic confusion, changed treatment strategies, and provided cascade testing for at risk family members. Affected family members may now benefit from targeted treatments, including angiotensin-converting enzyme inhibitors if micro-albuminuria occurs with the disease, as well as pre-emptive ophthalmology and audiology review.
This case highlighted two important issues: (a) the need for caution when analyzing VUS that potentially matches the patient phenotype, as these may be upgraded with time and (b) the need for structured re-evaluation of VUS in cases particularly where there is a high index of suspicion for genetic CKD, or when new data becomes available supporting the diagnosis. Studies have reported that up to 20% of VUS have been upgraded to likely pathogenic or pathogenic when re-evaluated. 10 As new information is continually emerging on genes and variants, systems to ensure structured re-evaluation of VUS are required to allow for the upgrade and or downgrade of a VUS over time.
Living kidney donation poses a unique challenge due to the time sensitive nature of the donation process and the irreversibility of the procedure. Overall outcomes in living kidney donors are excellent and comparable to outcomes in the general population. 17 Consensus guidelines are lacking on when genetic testing and how genetic results should be integrated into the donor assessment process. Increasingly data suggests that genetic testing can be considered in higher risk donors such as those with positive family history of CKD and those with concerning pre-donation features such as persistent proteinuria or hematuria or donors of younger ages who are biologically related to their recipient. 18 Current Canadian guidelines for living donation state that a diagnosis of AS precludes an individual from proceeding as a living donor, whereas a diagnosis of “thin basement membrane,” when all other testing is normal and when there is absence of any significant glomerular pathology can proceed with donation. 19 There is however ambiguity on whether potential donors who are heterozygous carriers of variants in the COL4A3 and COL4A4 genes, and therefore have a diagnosis of ADAS, with or without hematuria, should proceed with donation. Recently, a systematic review reported patients with ADAS due to COL4A3 and COL4A4 pathogenic variants with persistent microscopic hematuria. 20 They found that about 30% of patients developed CKD, 15% developed ESKD with a median age of onset of 50 years, and about 10% had extrarenal features. 20 This highlights the importance of a genetic assessment and detection of heterozygous mutations in the COL4A3 and COL4A4 especially when there is a positive family history of CKD and/or consideration as a donor, as this may increase the risk of developing CKD. Recent guidelines in the United States suggest that individuals with pathogenic heterozygous COL4A3 or COL4A4 variants and additional risk factors should not act as kidney donors because of the potential risk of kidney impairment after kidney donation. 1 Guidance is also lacking on how and when to incorporate genetic findings into the decision-making algorithm for living kidney donation. In this case, a pathogenic or likely pathogenic variant at the time of donor assessment may have added additional data on future risk of CKD post-donation which could aid in the informed decision-making process. Unfortunately, based on the information available at the time of pre-donation assessment, this variant was classified as a VUS, which is not a medically actionable finding.
This study was limited by the lack of biopsy on the patient to rule out CKD due to sarcoidosis. Although normotensive with normal calcium level, normal urinary calcium level, and no improvement in creatinine post-steroid therapy, it is entirely conceivable that renal sarcoid may be contributing to the kidney phenotype observed in this patient. However, with this case, we aim to highlight the possibility of a dual diagnosis and the importance of re-evaluation of genetic testing result in the context of new clinical features. We also want to highlight how the identification of a now likely pathogenic mutation in COL4A4 can support the diagnosis of ADAS not only in the index patient but also at-risk family members.
In summary, we show that re-evaluation of genetic data can lead to reclassification of genetic findings, which can have significant clinical implications for the patient and at-risk family members. This case supports the use of genetic assessment for both the diagnosis of ADAS in a patient with persistent haematuria and to counsel potential donors with persistent hematuria on the risk of developing CKD later in life post-donation. In accordance with previous data, we suggest a reanalysis period of 18 to 36 months, especially in high-risk patients. However, we acknowledge the potential limitations and ethical consideration pertaining to re-analysis in an individual who has already undergone living kidney donation. This case therefore highlights the importance of developing guidelines for both genetic testing and re-analysis of genomic data in living kidney donors as well as the need for further evidence on how this should be integrated into the living kidney donor assessment process.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: DMC’s involvement is funded by the Eugen Drewlo Chair for Kidney Research and Innovation at the Schulich School of Medicine & Dentistry at Western University, London, Ontario, Canada, the Academic Medical Organization of Southwestern Ontario (AMOSO) Innovation Fund, and the Department of Medicine Research Fund, London Health Sciences Centre.
Ethics Approval and Consent to Participate
Informed consent was obtained from the patient.