
Abstract
Backgound:
The introduction of the pneumococcal conjugate and polysaccharide vaccines have been valuable tools for combating invasive pneumococcal infection in children and healthy adults. Despite the available vaccination strategies, pneumococcal pneumonia and associated diseases continue to cause substantial morbidity and mortality, particularly in individuals with chronic disease and ageing populations. Next-generation pneumococcal vaccines will need to be highly immunogenic across patient populations providing both mucosal and systemic protective immunity. Mucosal immunization is an effective strategy for stimulating the immune response at the site of pathogen entry while increasing systemic immunity. In this study we utilized intranasal immunization with pneumococcal surface protein A (PspA), in combination with the mucosal adjuvant cholera toxin B (CTB), to characterize the immune components providing protection against S. pneumoniae challenge.
Methods:
Mice were immunized intranasally with CTB and PspA individually, and in combination, followed by lethal bacterial challenge with S. pneumoniae, strain A66.1. Animals were monitored for survival and tested for lung bacterial burden, cytokine production as well as S. pneumoniae-specific antibody titer in mouse sera. The primary immunological contributor to the observed protection was confirmed by cytokine neutralization and serum passive transfer.
Results:
The combination of CTB and PspA provided complete protection against bacterial challenge, which coincided with a significant decrease in lung bacterial burden. Increases in the T-helper (Th) 1 cytokines, interferon (IFN)-γ and interleukin (IL)-2 were observed in the lung 24 h post-challenge while decreases in proinflammatory mediators IL-6 and tumor necrosis factor (TNF)-α were also recorded at the same time point. The adjuvanted PspA immunization induced significant titers of S. pneumoniae-specific antibody in the serum of mice prior to infection. Serum adoptive transfer passively protected animals against subsequent challenge while IFN-γ neutralization had no impact on the outcome of immunization, suggesting a primary role for antibody-mediated protection in the context of this immunization strategy.
Conclusion:
Mucosal immunization with CTB and PspA induced a local cellular immune response and systemic humoral immunity which resulted in effective reduction of pulmonary bacterial burden and complete protection against S. pneumoniae challenge. While induction of the pleiotropic cytokine IFN-γ likely contributes to control of infection through activation of effector pathways, it was not required for protection. Instead, immunization with PspA and CTB-induced S. pneumoniae-specific antibodies in the serum prior to infection that were sufficient to protect against mucosal challenge.
Introduction
S. pneumoniae is associated with considerable morbidity and mortality especially in children and older adults. In addition to health concerns caused by bacterial pneumonia, S. pneumoniae frequently exacerbates lung conditions such as chronic obstructive pulmonary disease resulting in additional hospitalizations and public health burden. 1 Invasive pneumococcal disease is preceded by asymptomatic nasopharyngeal colonization mediated by bacterial interactions within the host mucosal niche. 2 Despite the importance of mucosal immunity in the prevention of pneumococcal disease, conventional vaccines are administered via intramuscular injections which often fail to induce mucosal immunity and often have limited efficacy in the highest risk populations. 3 Delivery of immunogens through the mucosal epithelia would be an ideal, minimally invasive alternative for protection against a mucosal pathogen such as S. pneumoniae. In addition, the close association of the mucosal epithelia with the underlying immune effector cells provides potential for induction of both mucosal and systemic adaptive immunity. 4
While intranasal immunization strategies are an attractive alternative, development of successful mucosal vaccines has proven difficult. One hurdle in this endeavor has been the lack of safe and effective mucosal adjuvants. Cholera toxin B (CTB), the nontoxic subunit of cholera toxin, has received attention as a potential adjuvant for its mucosal immunogenicity and affinity for the GM1-ganglioside receptor. The GM1 receptor is distributed on a variety of cell types, including the mucosal epithelia and immune cells such as macrophages, dendritic cells, and B-cells, allowing for rapid uptake through the mucosal barrier and enhanced interaction with immune effector cells. 5 Targeted and efficient binding to GM1 by CTB also allows for substantial reduction of administered antigen during the course of immunization. Studies by Bitsaktsis and colleagues have shown that uncoupled CTB administered intranasally with inactive Francisella tularensis enhances both humoral and cellular immune responses against subsequent bacterial challenge. 6 Development of vaccines against bacterial pathogens have attempted to utilize the immunogenicity of CTB by coupling bacterial subunits, such as Helicobacter pylori urease, to recombinant CTB resulting in a vaccine fusion protein which effectively induced urease-specific antibodies and reduced H. pylori burden in the stomach.7,8 In addition, recombinant CTB fusion proteins linked to an HIV-1 gp-41 epitope induced high-titer antibodies that neutralized viral transcytosis across the mucosal membrane, demonstrating the ability of CTB to stimulate an effective immune response in the context of varying pathogens. 9
The mechanisms of protective pulmonary immunity against S. pneumoniae are complex and remain poorly understood. The lung relies on innate cellular components such as alveolar macrophages and neutrophils, which will in turn support the development of antigen-specific T and B lymphocytes that can control bacterial proliferation in the lungs. 10 B-cell stimulation is a precursor to increasing antibody production which mediates immunologic protection against pneumococcal disease. While the current available vaccine regiments have decreased the number of invasive pneumococcal infections, such as septicemia and meningitis, the pneumococcal polysaccharide vaccine used in adults shows limited efficacy against all-cause pneumonia. 11 Evidence of the disease burden and emergence of nonvaccine serotypes emphasize the need for continued research and development of pneumococcal vaccines that induce long-lasting adaptive immunity with a strong protective response in the lung.
In the current study the mucosal adjuvant CTB was used in combination with the pneumococcal surface protein A (PspA) to test the efficacy of CTB at inducing an effective humoral and mucosal immune response when used as an adjuvant. PspA is a highly immunogenic surface protein that is expressed across most strains of pneumococci and has been well characterized as a potent vaccine antigen.12,13 Previous studies using intranasal immunization with PspA from clade 5 protected mice against bacterial challenge characterized by secretion of interleukin (IL)-17 and interferon (IFN)-γ in the lung and spleen. 14 Furthermore, a phase I trial in humans with recombinant PspA elicited post-immune sera samples that passively protected mice from fatal infection. 15 As an adjuvant, CTB has been used in combination with PspA to induce anti-PspA antibody in the sera and breast milk of pregnant mice which in turn protected offspring from infection in infancy, demonstrating the ability of CTB to effectively enhance the production of protective antibody. 16
In our study, administering PspA with CTB provided a significant survival advantage in mice, over PspA alone. Protection against mucosal challenge with serotype 3 clade 2 S. pneumoniae strain was accompanied by an increase in pathogen-specific antibody titer prior to infection as well as the Th1-type cytokine, IFN-γ, in the lungs of immunized mice following infection. However, the increased protection observed was, primarily, antibody dependent rather than T-cell mediated, demonstrating the importance of antibody production as a correlate of protection in adjuvanted mucosal immunization.
Materials and methods
Mice
C57BL/6 mice were procured from Jackson Labs (Bar Harbor, Maine, USA). Mice were housed in the animal facilities at Seton Hall University (South Orange, NJ) and provided with food and water ad libitum throughout the study. Mice used in our study were 6–12 weeks of age. All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee (South Orange, NJ).
Bacteria
The serotype 3 S. pneumoniae strain A66.1 was provided by Dr Gosselin (Albany Medical College, Albany, NY, USA). A66.1 was cultured in Todd-Hewitt Broth (Sigma-Aldrich, St. Louis, MO, USA), at 37°C until mid-log phase. The culture was then washed with phosphate-buffered saline (PBS) and resuspended in fresh broth containing 10% glycerol after which the stock was stored at −80°C until use.
Immunization and challenge experiments
Groups of five wild-type C57BL/6 mice were immunized intranasally with 20 µl of PBS, 1 µg of CTB (Sigma-Aldrich, St. Louis, MO), 5 µg of PspA (gift of Dr Snapper, Uniformed Services University of Health Sciences, Bethesda, MD), or CTB in combination with PspA. The immunization regimen was repeated on day 14 and 28 followed by i.n. challenge on day 42 with 1 × 106 colony-forming units (CFU) of live S. pneumoniae bacteria. Mice were monitored for 21 days following the challenge. The quantity of bacterial CFU administered was confirmed by culturing and counting the prepared inoculum on blood agar plates (trypticase soy agar with 5% sheep blood). The amount of PspA used was the dose that provided 50% protection in bacterial challenge studies.
Measurement of S. pneumoniae-specific antibody production
In order to detect the S. pneumoniae-specific antibody isotypes, enzyme-linked immunosorbent assay (ELISA) plates were coated in 1 × 107 CFU/well of live S. pneumoniae bacteria as previously described. 17 Coated plates were incubated overnight at 4°C and then washed four times with PBS containing 0.05% Tween-20. Washed plates were then blocked with superblock (Thermo Scientific, Waltham, MA) as indicated by the manufactures instructions. Serum samples collected 2 days before infection via submandibular bleed were added in duplicate as a two-fold serial dilution starting at 1:100, and incubated for 2 h at 37°C. Following incubation, plates were washed three times and an anti-mouse isotype-specific horseradish peroxidase conjugated secondary antibody was added for 1 h at 37°C [anti-immunoglobulin (Ig)G, anti-IgG2c, anti-IgG1 (Invitrogen, Carlsbad, CA)]. After 1 h incubation with secondary antibody the plates were washed three times and 3,3’,5,5’ tetramethylbenzidine (TMB) substrate solution was added as per manufacturer instructions, the sample optical density (OD) was read at 650 nm on a microplate reader. The antibody titer was determined as the reciprocal of the highest dilution that provided a reading two-fold greater than the average OD generated in the PBS treatment group.
Quantification of bacterial burden and cytokine measurement
Mice were immunized and challenged as described above. At 24 h post-infection the mice were sacrificed using CO2 administration followed by cervical dislocation. Lung tissue was collected aseptically in PBS containing protease inhibitor (Thermo Scientific, Waltham, MA, USA) and subjected to mechanical homogenization using an Omni tissue homogenizer. Supernatants were then diluted 10-fold and 10 µl of each dilution was spotted in duplicate on blood agar plates. Plates were incubated at 37°C for 24 h and monitored for growth. The remaining tissue suspension was centrifuged at 12,000 × g for 20 min and the supernatant was removed and frozen at −20°C for cytokine analysis. Cytokine levels were assayed using mouse ELISA Max kits (Biolegend, San Diego, CA, USA).
Serum adoptive transfer
Groups of five mice were immunized as described above for use in the passive transfer experiment. At 2 weeks after the second boost (day 42), blood was collected via cardiac puncture. Blood samples were allowed to clot for 30 min at room temperature and then spun at 10,000 × g for 90 s to separate the liquid component, the resulting serum from the same experimental group was pooled and stored at −20°C. Sera preparations were heated at 55°C for 30 min to inactivate complement and then administered (150 µl per mouse) via intraperitoneal injection 24 h prior to A66.1 challenge.
Cytokine neutralization
To determine the importance of IFN-γ in the CTB-protective immune response, IFN-γ was neutralized in two groups of five mice immunized with CTB and PspA as described above. To neutralize the cytokine 500 µg of LEAF™ purified IFN-γ antibody (clone XMG1.2) or LEAF™ purified rat IgG1, κ isotype control was administered in two consecutive daily doses of 250 µg by i.p. injection prior to i.n. infection with A66.1.
Statistical analyses
Statistical analysis of survival studies was conducted using the log-rank (Mantel–Cox) test. Statistical data for bacterial burden, cytokine analysis and antibody titers were assessed using a one-way analysis of variance or the unpaired, two-tailed Student’s t-test. Statistical analysis and graphs were performed using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA, USA).
Results
Mucosal immunization with CTB and PspA decreases lung bacterial burden and enhances protection against S. pneumoniae
In the beginning of our study, we sought to establish the PspA immunization amount that provided approximately 50% protection in mice against an LD100 S. pneumoniae challenge. Using a two-boost regime (days 14 and 28), we found that 5 μg of PspA per immunization (intranasally) provided 40–60% protection in mice (data not shown). We then proceeded to test the adjuvant efficacy of CTB in promoting a protective immune response against pneumococcal bacterial infection. To this end, mice were immunized intranasally with 5 μg of PspA mixed with 1 μg of CTB, or PspA alone, on our two-boost established schedule. Intranasal administration of CTB in conjunction with PspA resulted in complete protection against lethal bacterial challenge with serotype 3 S. pneumoniae (strain A66.1). As expected, only 40% of animals that received PspA alone were protected from bacterial challenge while CTB provided no protection when administered independently of PspA (Figure 1a). The bacterial burden was also significantly decreased in the lungs of mice that received the adjuvanted PspA. Lung tissue was collected 24 h post-challenge, homogenized and plated on blood agar plates. The lungs of animals immunized with PBS, PspA or CTB alone had six-fold higher bacterial load compared with mice immunized with CTB and PspA (Figure 1b). High levels of proliferating bacteria in the lung are indicative of an uncontrolled infection which can progress into septicemia. Co-administration of the mucosal adjuvant with PspA provided sufficient stimulation to clear the infection in the lung effectively, thus limiting systemic disease.
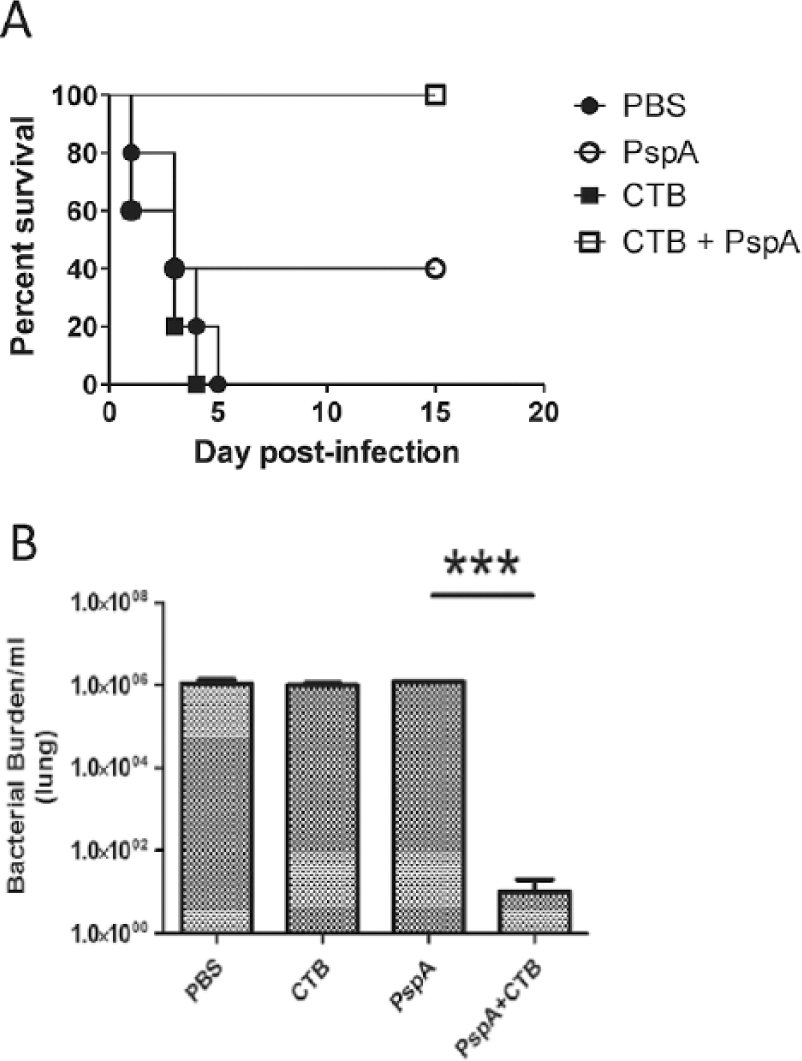
Intranasal administration of PspA with CTB reduces bacterial burden and protects against mucosal challenge with S. pneumoniae. Mice immunized i.n. with PBS, PspA (5 µg), CTB (1 µg), or PspA with CTB, and boosted twice on days 14 and 28, were challenged with live S. pneumoniae, strain A66.1 (1 × 106 CFU) 2 weeks after the second boost (day 42) and the survival was monitored for at least 15 days after infection (a). Bacterial burden in the lungs of immunized mice was measured for each group 24 h post-infection by plating on blood agar plates (b). A total of eight mice were used per group and this is representative of three experiments.
Use of CTB as a mucosal adjuvant drives production of pathogen-specific antibodies
To assess the level of S. pneumoniae-specific antibodies induced by intranasal immunization with the adjuvanted protective pneumococcal antigen, PspA, we collected sera from immunized mice, via submandibular bleed, 2 weeks after the second boost. The titer of antibody isotypes (IgG, IgG1, IgG2c, and IgA) was measured by ELISA using S. pneumoniae-coated plates. Intranasal immunization with CTB and PspA significantly increased the amount of pathogen-specific total IgG, IgG1, and IgG2c in mouse sera, compared with PspA alone. The significantly higher levels of both IgG2c and IgG1 antibody titers indicated a mixed Th1 / Th2 response (Figure 2). Surprisingly, we were not able to detect any IgA-specific antibody titers despite this being the predominant isotype found in the mucosa. Nevertheless, the significant increase in pathogen-specific antibody secretion observed in mice immunized with adjuvanted PspA demonstrates the ability of CTB to generate pathogen-specific humoral immune responses when used as a mucosal adjuvant.

Characterization of S. pneumoniae-specific antibodies in the sera of mice immunized with PspA adjuvanted with CTB. Mice were immunized as previously described. After 2 weeks from the second boost, S. pneumoniae-specific serum titers of IgG (a), IgG2C (b), and IgG1 (c) were detected by isotype-specific ELISAs. Each time point is the average of five mice and the data are representative of three independent experiments (*, p < 0.05 and **, p < 0.01, n = 5).
Levels of adaptive Th1-type cytokines IFN-γ and IL-2 are increased in the lungs of mice immunized with CTB and PspA following bacterial challenge
In order to determine the contribution of cellular immunity to the respiratory response mediated by immunization, cytokine production in lungs of immunized mice was profiled 24 h following bacterial challenge with S. pneumoniae. Levels of the Th1 cytokines, IL-2 and IFN-γ, were significantly increased when PspA was co-administered with CTB (Figures 3a and 3b). Both these cytokines are produced by Th1 cells and can sustain the activation and proliferation of CD4+ T-cells, 18 indicating the ability of CTB to elicit a T-cell-dependent immune response during bacterial infection when used as a mucosal adjuvant. Interestingly, the Th2 type cytokine, IL-4, was found to be elevated in the lungs of all mice after infection (Figure 3c). However, measuring the levels of IL-4 prior to infection, indicated a significant increase in the lungs of mice immunized with CTB and PspA, compared with mice immunized with PspA alone, thus justifying the higher levels of the S. pneumoniae-specific IgG1 isotype previously measured (Figure 2c and data not shown). While the induction of T-cell cytokines can be paramount in the resolution of bacterial infections, prolonged inflammatory mucosal responses, such as in the lung, must be regulated to avoid tissue injury. To this end, we measured the proinflammatory cytokines, IL-6 and TNF-α, in the lungs of immunized mice 2 days after S. pneumoniae challenge. Mice immunized with CTB and PspA had significantly lower levels of these cytokines which is indicative of resolution of infection (Figures 3d and 3e).

Utilization of CTB as a mucosal adjuvant modulates cytokine production in the lungs of immunized mice. Mice were sacrificed 24 h post-challenge and lungs were harvested and homogenized for cytokine profiling by ELISA. Levels of IFN-γ and IL-2 were increased in mice immunized with CTB in combination with PspA (a and b) while infection caused elevated levels of IL-4 independent of immunization (c). Proinflammatory cytokines IL-6 and TNF-α were decreased in mice immunized with the adjuvanted PspA (d and e, respectively). Data represent the average of five mice and is representative of three experiments (*, p <0.05 and **, p < 0.01, n = 5).
Antibodies but not IFN-γ are required for protection in PspA plus CTB-immunized mice
Intranasal immunization of mice with PspA and CTB showed evidence of enhanced humoral and cellular responses. In order to investigate whether both aspects of the immune response are necessary for protection against S. pneumoniae challenge when CTB is utilized as a mucosal adjuvant, we performed serum transfer and IFN-γ neutralization experiments. To evaluate the protection conferred by vaccine-induced antibodies, sera was obtained from immunized mice for adoptive transfer. The serum was heat inactivated to neutralize complement and transferred intraperitoneally to naïve animals (150 µl/mouse) 24 h prior to bacterial infection. Animals that received sera from the CTB plus PspA immunized mice had an 80% survival rate compared with 20% survival in animals treated with the PspA sera alone. As expected, the transfer of control sera from PBS immunized mice provided no protection against challenge (Figure 4a).
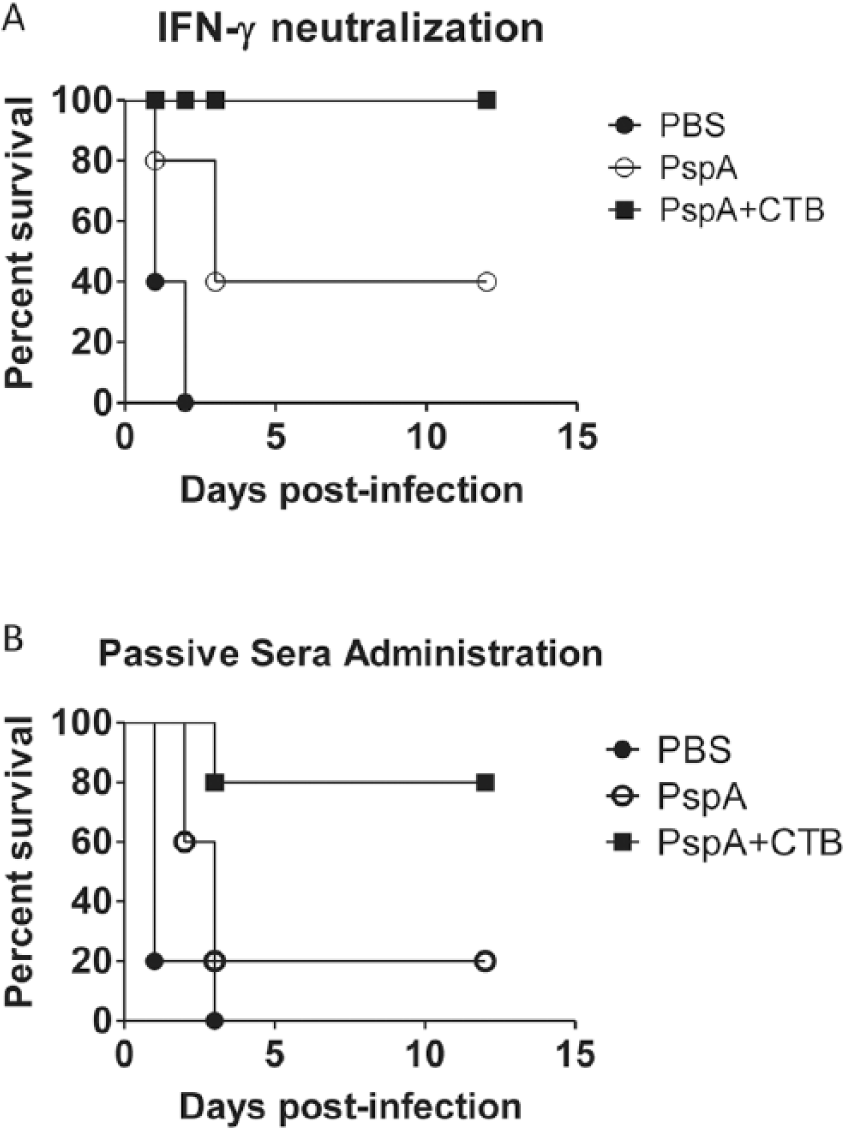
Humoral immunity is the primary contributing factor to CTB-induced protection. After 2 weeks from the second boost, blood was pooled from the respective immunized mouse groups and processed for serum. Serum was administered intraperitoneally (150 µl per mouse) 24 h prior to S. pneumoniae challenge. Animals receiving sera from the CTB + PspA immunized group were passively protected from mucosal infection with S. pneumoniae (a). Immunized mice were treated with neutralizing antibody or an isotype control prior to infection. Neutralizing IFN-γ did not affect the protection observed when mice were immunized with the adjuvanted PspA (b). A total of five mice were used per group and the data are representative of two experiments.
To evaluate the importance of IFN-γ and thus, that of T-cell responses, immunized mice were treated with an IFN-γ-neutralizing antibody (clone: XMG1.2) through i.p. injection and then challenged with a lethal dose (LD100) of S. pneumoniae. To account for any antibody-induced toxicity an isotype control was used. In contrast to the results observed with serum transfer, neutralization of IFN-γ prior to bacterial challenge did not affect the protection induced via immunization with CTB and PspA (Figure 4b). As documented in the initial survival study, 100% of immunized animals were protected against bacterial challenge regardless of their ability to utilize IFN-γ.
Discussion
Localized respiratory immunity is essential for protection against S. pneumoniae infection and preventing development of systemic disease. In our study, utilization of the mucosal adjuvant CTB in combination with the protective pneumococcal protein antigen PspA significantly amplified antigen-specific antibodies prior to infection and Th1-associated cytokines in the lungs, resulting in protection from pneumococcal infection. While the presence of a localized cellular response is likely a contributor to containing infection, antibody-mediated immunity was paramount to survival outcomes in mice receiving CTB and PspA as demonstrated by our serum transfer experiments. The presence of S. pneumoniae-specific antibodies induced by vaccination, can elicit protection through blockage of bacterial adhesion and complement independent opsonization. IgG class antibodies are effective in both these immunological processes; however, the important mucosal antibody isotype, IgA, was not detected in any significant amounts in our study. Furthermore, serum antibody inhibition in vitro significantly inhibits S. pneumoniae uptake by phagocytic cells compromising bacterial clearance, which suggests the presence of protective antibodies most likely contributed to the reduced bacterial burden observed in the adjuvanted immunization group. 19
In addition to the high titers of S. pneumoniae-specific antibodies observed pre-infection, immunization of mice with adjuvanted PspA increased IFN-γ and IL-2 cytokines while it decreased the proinflammatory cytokines IL-6 and TNF-α within 24 h after bacterial challenge. Both TNF-α and IL-6 are critical early cytokines produced by alveolar macrophages in the first minutes to hours following pathogen recognition and defense. 20 This innate proinflammatory response is initially a localized occurrence which serves to orchestrate the immune cascade through neutrophil recruitment and adaptive mechanisms such as T-cell polarization and B-cell stimulation. Excessive production of TNF-α can exacerbate neutrophil activation and instigate lung damage. 21 Similarly, progressive increases in serum and lung IL-6 24 h post-infection is correlated with bacteremia and death in mice infected with type III S. pneumoniae. 22 Early cessation of pulmonary inflammation is a beneficial outcome of CTB adjuvant to minimize tissue damage and expedite recovery following bacterial exposure.
In the current study we observed a decrease in the innate proinflammatory mediators in parallel with a decrease of lung bacterial burden. Additionally, while these acute phase cytokines were declining, indicators of the long-lasting adaptive response were increasing in the form of Th1 signature cytokine IFN-γ and IL-2. Th1 subsets mediate post-vaccination protection through the production of cytokines like IFN-γ and IL-2 that stimulate CD8+ cytotoxic T-cells, natural killer cells, and innate lymphoid cells. 23 Additionally, IFN-γ produced by Th1 cells at the site of infection can activate macrophage to secrete factors such as metalloproteinases and nitric oxide which promote phagocytic killing of invasive bacteria. 24 Classic Th1 cells differentiate from CD4+ T-cells when the T-cell receptor (TCR) is engaged with its cognate antigen that has been loaded onto the major histocompatibility complex class II molecules on the surface of antigen-presenting cells (APCs). The distinct T-cell lineage subset is defined by factors such as the costimulatory molecules on APCs, the local cytokine environment, and TCR signal intensity. Under fixed antigen concentrations adjuvants can influence T-cell polarization through APC-induced changes in the cytokine environment and expression of costimulatory molecules such as CD80 and CD86 that amplify the intensity of TCR engagement. 25 To this point, studies from our lab have shown that CTB increases the expression of CD80 and CD86 on murine macrophage in vitro, which could influence the differentiation of naïve CD4+ Th cells to Th1 lineage. 26
Classic Th1 cell differentiation also relies on secretion of IFN-γ by cells of the innate immune system such as neutrophils, which produce IFN-γ early during the inflammatory response to bacterial pneumonia. 27 IFN-γ production in the lung can contribute to containing infection through promoting bacterial clearance by neutrophil mediated host-protective responses. Specifically, the production of neutrophil extracellular traps, an important bactericidal mechanism is believed to be regulated by IFN-γ. 28 The role of IFN-γ as a neutrophil activating cytokine and promoter of Th1 differentiation suggests the benefit of a timely IFN-γ response, where early secretion is advantageous for engagement of the innate phagocytic cells and regulation of Th1 adaptive cell-mediated immunity, while continued IFN-γ release could be detrimental to recovery. 29
While IFN-γ is a functionally diverse cytokine that contributes to both innate and specific immunity, the protection afforded to animals receiving CTB and PspA was not found to be dependent on IFN-γ as indicated by our cytokine neutralization studies (Figure 4). Instead, humoral immunity drove survival outcomes of animals challenged with S. pneumoniae. Humoral defense is especially important in the context of biological PspA which has been shown to inhibit complement activation and C3 deposition during the course of infection resulting in septicemia.30,31 Antibodies against PspA can overcome complement inhibition induced by PspA, and increase C3 deposition on the surface of invading S. pneumoniae. 32 All three pathways of the complement system rely on C3 deposition to activate the complement cascade which has been well described as an essential component for identification and destruction of invading pneumococci.33,34 The role of humoral immunity in increasing the amount of complement bound to the bacterial surface could explain why CTB and PspA were unable to elicit protection against S. pneumoniae strain 6303 which expresses PspA from a different family than our challenge strain A66.1 (data not shown). Antibodies directed against PspA have shown to be cross-reactive against strains expressing PspA within the same family, with limited reactivity across different families. 35 Furthermore, survival against challenge with alternate strains of S pneumoniae has been shown to correlate with the amount of complement deposition induced by anti-PspA antibodies reinforcing the necessity of cross-reactive antibodies for protection against varying strains regardless of the immune enhancements observed with CTB. 36
While IFN-γ may have not been essential for protection against bacterial challenge the combination of humoral and pulmonary immunity induced by CTB provided both a local and systemic response to effectively overcome S. pneumoniae infection. The ability of CTB to modulate inflammatory signals while stimulating adaptive cytokine and antibody production is a valuable tool which will need to be emulated in future vaccine strategies to provide more broadly protective vaccine regiments for those still ask risk from S. pneumoniae infection.
Footnotes
Acknowledgements
We would like to thank Dr E Gosselin (Albany Medical College, Albany, NY, USA) for providing the S. pneumoniae type III (A66.1 strain) and Dr C Snapper (Uniformed Services University of the Health Sciences, Bethesda, MD, USA) for providing the recombinant PspA for our experiments.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by Seton Hall University start-up research funds.
Conflict of interest statement
The authors declare no conflicts of interest in preparing this article.