
Abstract
Summary
The association of intestinal dysbiosis with the pathogenesis of inflammatory bowel disease has been well established. Besides bacteria, microbiota comprises yeasts, archaea, protists and viruses, neglected actors in inflammatory bowel disease-associated microbiota. In the past, a great limitation in studying microbiota composition was the low sensitivity of sequencing technologies and that few computational approaches were sufficient to thoroughly analyse the whole microbiome. However, new cutting-edge technologies in nucleic acid sequencing, -omics analysis and the innovative statistics and bioinformatics pipelines made possible more sensitive and accurate metagenomics, ultimately identifying novel players in intestinal inflammation, including prokaryotic and eukaryotic viruses, that together form the gut virome. The discovery of peculiar inflammatory bowel disease-associated microbial strains will not only shed new light on inflammatory bowel disease aetiogenesis, they may also support the development of novel therapeutic strategies not merely treating symptoms, but precisely counteracting the primary cause of chronic intestinal inflammation.
Introduction
During birth and over the first days of life, the gastrointestinal (GI) tract starts being colonized by microbial species that constitute the gut microbiota, where they coexist in a dynamic equilibrium, actively interacting with the host. 1 In newborn babies, such a ‘dynamic consortium’ composition depends on either the delivery mode 2 or way of feeding 3 and is inherited from the mother, whose dominant strain is transferred to the child and transiently populates the gut during birth. Within the first 3 years, the child continues gaining microbes from distinct maternal sources.4,5 The microbiota changes until adulthood when it reaches more stability, 6 at least in healthy conditions. 7 Nevertheless, environment, diet, daily habits and antibiotic treatments affect this stability, modulating microbiome composition throughout the lifespan and eventually influencing the host in health and disease.8,9
The gut microbiota has favourable effects on our health. For example, species belonging to the Bifidobacterium and Lactobacillus genera have been shown to protect enterocytes from an acute inflammatory response counteracting enteropathogen infections. 10 Moreover, the microbiota ensures gut homeostasis by modulating immune functions and contributes to the absorption of nutrients from food, sometimes cooperating with the host for their metabolism. 6 Therefore, interruption of the balance between the gut microbiota and the host may lead to pathological conditions such as inflammatory bowel disease (IBD). 11
IBD is a class of diseases comprising Crohn’s disease (CD) and ulcerative colitis (UC), characterized by an exacerbated immune response due to gut epithelial barrier disruption and bacterial translocation from the lumen to the mucosal tissue.12,13 This leads to the activation of both innate and adaptive immune responses, ultimately failing to be resolved and leading to chronic inflammation (Figure 1).
12
Therefore, intestinal microbiota play a pivotal role in IBD etiopathogenesis and understanding the mechanisms through which it intervenes and affects this disease are essential to discover novel therapeutic approaches.
Microbiota in inflammatory bowel disease (IBD). Healthy gut lumen is populated by different microbial species, including viruses, bacteria, yeasts, protists and archea (left panel). In IBD (right panel), epithelial barrier disruption leads to the translocation of microbes that colonize mucosa. This leads to the activation of both innate and adaptive immune responses, ultimately failing to be resolved and leading to chronic inflammation.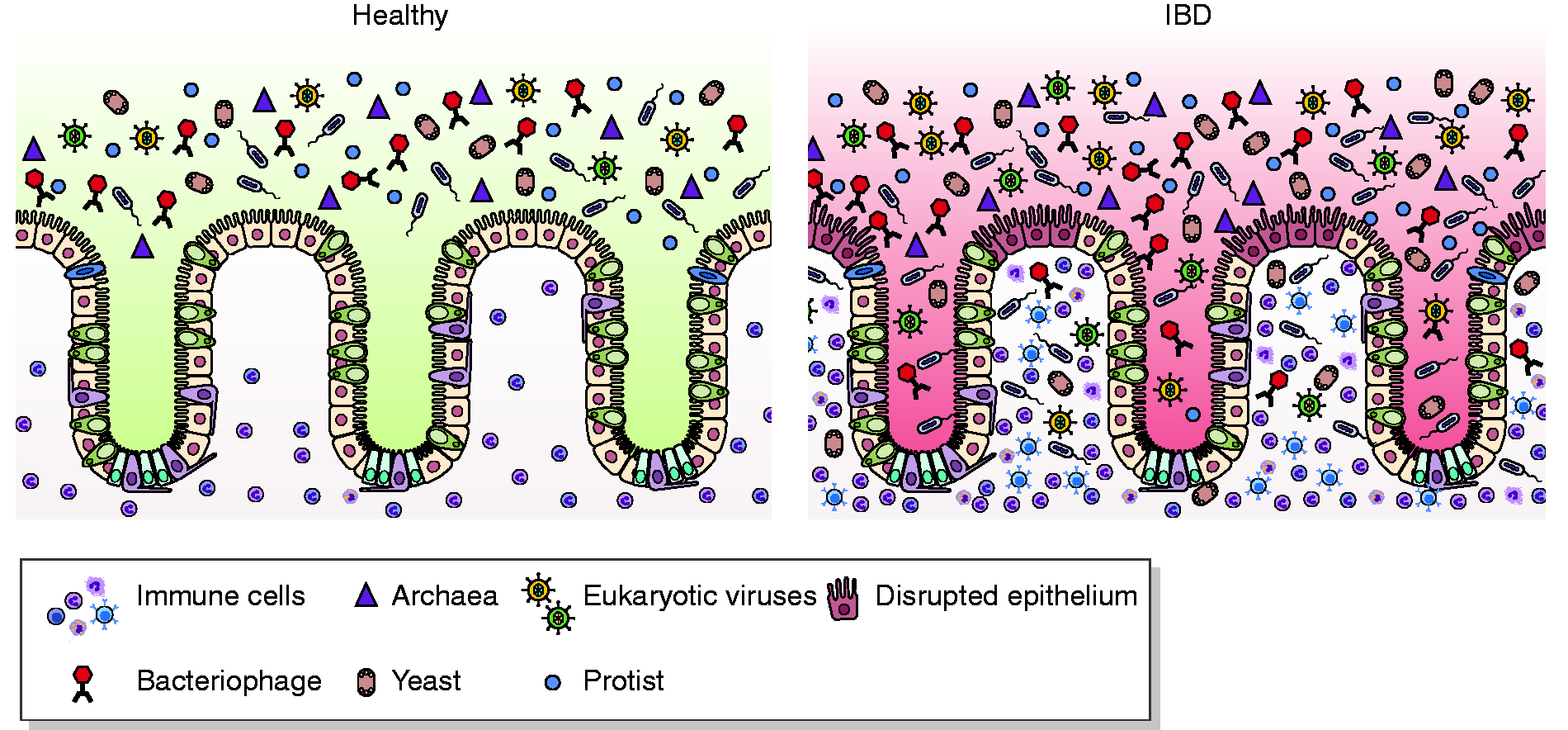
Here we provide an overview of the techniques for microbiota-related studies and how the microbiota may affect IBD pathogenesis, with a focus on the virome as a possible trigger for intestinal inflammation.
Next-generation sequencing: The breakthrough for microbiota discovery
Although consistent attention was given to the microbiota contribution to IBD pathogenesis in the past, more exhaustive works elucidating this have been published only recently, after next-generation sequencing (NGS) was established. 14
Although bacterial culture-based systems allowed highly accurate, extensive definition of bacterial genus and species abundance, 15 they were time consuming because of protocol optimization and low throughput. Moreover, many entities populating the gut microbiota, such as viruses, archaea, protists and fungi, were neglected because of the impossibility of creating a dedicated composite culture condition in vitro sustaining the growth of different microorganisms at the same time.14,16,17
The advent of system biology posed the first milestone for the composite and detailed study of the microbiota. In fact, the ‘-omics era’ made possible the unbiased analysis of members of the microbiota, their genes, transcripts, metabolites and proteins isolated from biological samples. 14
First studies exploiting NGS defined the bacterial diversity within microbiota by prokaryotic 16S rRNA gene-targeted sequencing (metataxonomics). 18 These are highly evolutionary conserved genes displaying hypervariable regions that allow researchers to analyse differential microbial enrichment within a biological sample.
A more unbiased approach, exploiting untargeted sequencing, was shotgun metagenomics, which explores the whole DNA in an environment. 19 This different from metataxonomics, where barcoded primer sets target the 16S variable regions; metagenomics is based on the fragmentation of DNA, its barcoding with random primers, sequencing and analysis via dedicated bioinformatic pipelines. 20
Metagenomics provided investigators with high throughput data describing complex microbiomes. In fact, such an approach covers the investigation of different kingdoms of the microbiota and allows a more comprehensive analysis of its composition. 21
The great limitation of metagenomics is represented by the analysis of DNA sequences only, thus ignoring RNAs. So far, metatranscriptomics has embodied a unique and useful approach to overcome this issue. In fact, by analysing the RNA composition of a biological sample (such as gut mucosa) and exploiting cutting-edge NGS technologies, the analysis of the whole transcriptome may point out new microbial entities (such as RNA-based viruses) 22 that represent an active part of the microbiota.
It is noteworthy that metataxonomics, metagenomics and metatranscriptomics share the same limitations: 23 1) they must rely on accurate databases featuring the different genomes and their annotations, otherwise the analysis remains elusive and may lose some important information; 14 2) they need to be performed on purified RNA and DNA samples, but sometimes yields are not sufficient to cover less represented entities of the microbiota; moreover residual DNA and RNA molecules from the host may remain in the sample after purification, producing misleading results; 14 3) the depth of the sequencing has to be very high, mainly for metatranscriptomics, to ensure reliable results, 24 which might be costly; and 4) the current statistical approach does not contemplate the biological system’s complexity and is limited by the assumption that predictor variables (microbial species) are independent of each other. Such an assumption cannot be applied to the complex human gut microbiota. 14 By contrast, machine learning algorithms, including neural networks, support vector machines and decision trees, 25 may allow decoding of the gut microbiota intricacy by exploiting pattern recognition, which does not require the identification of predictor variables in advance, also managing a large volume of data. 14
Remarkably, a bioinformatics and computational approach alone will not be able to elucidate the microbiota-associated mechanisms underlying IBD pathogenesis and experimental works are needed to demonstrate their causality. 26 Nevertheless, although NGS technology needs improvements and in silico data need to be empirically demonstrated, this field is becoming very promising. In fact, the advances in NGS are very quick so in few years all limitations might be overcome and more accurate conclusions may come sooner.
Gut microbiota in intestinal chronic inflammation: Evidence from human and animal studies
More commonly, the human intestine is colonized by bacteria.22,27 With more than 1000 species, the main bacterial phyla residing in the gut are Bacteroidetes and Firmicutes, followed by Actinobacteria and Proteobacteria. 28
Another consistent but neglected proportion of the gut microbiota is made up of fungi, archaea, viruses and protists. 29 Because of their coexistence in the same area, these microorganisms are supposed to actively interplay with each other through defence while competing for nutrients, or with cooperation while growing synergistically. 30 Such a relationship subsidizes gut homeostasis maintenance, performing several roles in health and disease31,32 that will be discussed in the following sections.
Bacteria
Substantial shifts in the overall microbiota composition have been associated with IBD etiopathogenesis, whose most relevant feature is the reduction of bacterial diversity in the microbiota structure and higher levels of Proteobacteria, coupled with lower proportions of Bacteroidetes and Firmicutes. 33 Among Firmicutes, a decrease in the Clostridium leptum groups, including the Faecalibacterium prausnitzii, has been reported in many studies.34,35 Of note, F. prausnitzii produces butyrate, 36 which is known to have beneficial effects for health.37–39
Another bacterium characterizing the IBD-associated microbiota is the Mycobacterium avium subspecies paratuberculos, detected in CD mucosal samples. 40 However, administration of antituberculosis drugs to CD patients was not effective. 41 Also, adhesive-invasive Escherichia coli (AIEC) was found to colonize the ileal mucosa of CD and correlate with disease location, activity and postoperative recurrence. 42 AIEC also replicates in macrophages stimulating TNFα production. 43
Whether IBD-associated intestinal microbiota changes are a cause or result of this disease is still debated. In this regard, the roles of some bacterial species have been explored to elucidate their causative effects in experimental colitis. For example, in T-bet−/− x Rag2−/− transgenic mise, used for spontaneous colitis investigation, Proteus mirabilis and Klebsiella pneumoniae abundances correlated with colonic inflammation symptoms in terms of histologic colitis score, 44 whereas in Il10-/- mice Bilophila wadsworthia, normally a minor component of the gut commensalism, was associated with colitis development. 45 More interestingly, Fusobacterium varium (FV) isolated from the colonic mucosa of UC patients displayed the capability of disrupting the epithelial barrier in mice, 46 raising the question of whether some microbiota components may trigger colitis. In this regard, the FV-contrasting combination therapy showed efficacy in UC patients, further supporting the possible pathogenic role of this microbial entity in chronic inflammation. 47
Fungi, archaea and protists
The gut mycome, made of fungal species, normally colonizes the GI of healthy subjects, affecting the immune system and producing specific metabolites. 48 Some may be beneficial, such as Saccharomyces cerevisiae, used as a probiotic to treat GI disorders. 49 A couple of years ago, a study pointed out a decrease in mycome alpha diversity, associated with bacterial dysbiosis, in patients with UC and, to a lesser extent, in patients with CD. 50 Likewise, Candida tropicalis was linked to CD bacterial dysbiosis and the presence of circulating anti-Saccharomyces cerevisiae antibodies correlated with its abundance. 51 Another study showed the fungus Malassezia restricta, abundant in CD patients, was linked to the IBD-associated polymorphism in the CARD9 gene, through which it triggered the innate inflammatory responses and worsened colitis in mice. 52 Very recently, other evidence has emerged from the systematic review conducted by Stamatiades and colleagues, depicting Candida species as the main fungal infection occurring in patients with IBD in both the respiratory and GI tract, soon after treatment commencement (mainly anti-TNFα), causing also serious complications. 53
Within the microbiota, up to 10% of all anaerobes are methanogenic archaea, with Methanobrevibacter smithii the most predominant in almost every human subject. 54 Although some evidence suggested archaea-dependent positive effects on human health, 55 several studies discuss whether archaea are detrimental for health by promoting pathogenic bacterium growth. 56 In this regard, the higher prevalence and immunogenicity of Methanosphaera stadtmanae are being discussed in association with the development of inflammatory conditions, including IBD. 57
Among enteric protozoans colonizing the human gut, Blastocystis is one of the most common unicellular eukaryotes detected in human stools 58 and new findings support the protective role of Blastocystis as a common constituent of the gut microbiota. In this regard, Blastocystis-colonized patients were shown to exhibited a higher abundance of Clostridia class and Ruminococcaceae and Prevotellaceae families, whereas Enterobacteriaceae were enriched in Blastocystis-free patients, suggesting Blastocystis colonization was not associated with the colitis-specific gut dysbiosis but with a healthy gut microbiota. 58 Recently, microbial richness and diversity were found to be linked to Blastocystis prevalence and subtype variation; its prevalence was reduced in patients with IBD, 59 further supporting the concept that this protozoan may be a common constituent of healthy gut microbiota.
All these studies illustrate fungi, archaea and protists as microbiota-specific components that participate in IBD pathogenesis. Nevertheless, their mechanistic roles in regulating chronic gut inflammation-associated events need to be further investigated by other explorative studies to dissect the complexity of the mutual interactions with each other and with the human host.
Virome: The neglected actor
Another large portion of the gut microbiota is composed of viruses, infecting both prokaryotic and eukaryotic cells and forming the gut virome. The coexistence of bacteria and viruses within the gut has increasingly attracted interest so that some studies are dedicating more attention to the role viruses play in gut homeostasis and pathogenic conditions. 60
The human gut virome is a mixture of three classes, including a bacteriophage core shared by more than 50% of people, another core found in 20–50% of the population and a set of rarely shared bacteriophages. 61 Prokaryotic-infecting viruses are 10-fold more abundant than bacteria, 62 thus meaning their existence within the microbiota strongly contributes to the modification of the different proportions of bacterial strains. This occurs by lysis of prokaryotic cells that live within the intestine or by genome integration, which can change either the surface structures of bacteria, thus affecting the bacteria-host interactions, or the bacterial fitness and phenotype, conferring antibiotic resistance, ability to produce toxins or increase energy yield.63–65 Therefore, gut phages are not only predators, but can help their host by providing genes that confer a competitive advantage in the ecosystem in which they both live. 64
On one hand, phages are closely connected to bacterial resilience and function, encoding genes necessary for bacterial mutualism with the host,9,60,66 but on the other hand, phage-encoded virulence factors may promote pathogenic events.67,68 One of the pioneer studies delineating gut virome dysbiosis as a condition associated with IBD pathogenesis showed the increased abundance of Clostridiales-, Alteromonadales- and Clostridium acetobutylicum-infecting phages, as well as more abundance of Retroviridae family in subjects with IBD by comparison with healthy subjects. 69 Another study performed on children showed the Caudovirales order to be more represented in IBD patients, 70 whereas recently Zuo and colleagues observed that a Caudovirales bacteriophage expansion, coupled with a decrease in their diversity, richness and evenness, occurred in UC patients and this signature was directly correlated with intestinal inflammation. Additionally, Escherichia phage and Enterobacteria phage were enriched in UC mucosa in comparison with the control. Notably, the UC virome showed abrogation of diverse viral functions, whereas functions of bacteriophages associated with host bacteria fitness and pathogenicity were enriched in UC mucosa. 71
The possible causative effect of bacteriophages in promoting chronic inflammation was recently highlighted by a study describing their ability to impact on the gut immune response in germ-free mice. Herein, Lactobacillus, Escherichia, and Bacteroides bacteriophages and phage DNA induced IFN-γ production via TLR9. Moreover, by increasing bacteriophage levels, colitis was exacerbated via TLR9 and IFN-γ.
Finally, the gut phage composition differs between UC and CD patients. In fact, elegant profiling of the human gut virome of stools from patients with CD and UC demonstrated not only that the virome structure was disease and cohort specific, but also that its variations contributed to intestinal dysbiosis. 72
Beside phage-associated pathogenesis, a therapeutic approach exploiting bacteriophages for the treatment of intestinal inflammation has been proposed. In fact, it has been shown that mice administered with a cocktail of bacteriophages benefited from a significant decrease in faecal AIEC number and in the adherent flora. Strikingly, a single dose of the cocktail not only reduced Dextran Sulfate Sodium (DSS)-induced colitis symptoms in mice but also targeted AIEC in homogenates of CD ileal biopsies. 73 The efficacy of a bacteriophage cocktail against AEIC in CD patients is under investigation in a clinical trial (www.clinicaltrials.gov; NCT03808103).
Eukaryotic viruses, which start colonizing gut mucosa early in life, belong to Adenoviridae, Anelloviridae, Astroviridae, Parvoviridae, Picornaviridae and Picobirnaviridae families and their richness increases with age. 74 These viruses may either lead to symptomatic manifestations, or they may remain latent for long in healthy people, also exerting beneficial effects.22,75–77
Similarly to phages, eukaryotic virome dysbiosis was associated with IBD pathogenesis,23,27,64 because eukaryotic-targeting viruses integrate into the human genome and may influence the physiological state of intestinal cells.22,64,76 Metagenomic analysis on a large cohort of UC patients showed the eukaryotic Pneumoviridae family was more enriched in UC patients than in controls, whereas the eukaryotic Anelloviridae family was higher in controls than in UC patients. 71 By contrast, another study reported that a small cohort of UC and CD patients displayed higher Herpesviridae family levels in comparison with control subjects. 78 Such a discrepancy might be due to the small sample size, thus not highlighting other slight differences between healthy and IBD-associated virome composition.
The most investigated eukaryotic viral entities possibly triggering intestinal inflammation are the cytomegalovirus (CMV) and the Epstein-Barr virus (EBV). Both CMV and EBV, members of the Herpesviridae family acquired early in life, are usually asymptomatic and may remain latent for the entire lifespan of healthy people. 77 However, to date, their involvement in IBD etiopathogenesis has yet to be elucidated as their reactivation may result from immunosuppression or stressing conditions common in IBD patients and therefore they might be bystanders instead of real triggers for this disease.
Hence, the effective role of virome in IBD aetiogenesis has yet to be discovered, even if some evidence depicting eukaryotic viruses as potential initiators of intestinal inflammation are available. In fact, in the Il10KO model of spontaneous colitis, the Norovirus infection was discovered as a potent colitogenic factor, strongly depending on the presence of enteric microbiota. 79 Similarly, results from IBD-susceptibility gene Atg16L1HM mouse models demonstrated that Norovirus infection contributes to the development of intestinal inflammation. 80 Both studies thus showed the synergistic effect between genetic background and Norovirus infection as a precipitator of intestinal inflammation, speeding up colitis development.
Notably, these studies emphasized enterotropic viruses, normally localized at the level of the GI. By contrast, a recent study exploiting metatranscriptomic pipelines showed that eukaryotic virus RNAs, with a physiological hepatic tropism, were detected within intestinal mucosa of a large cohort of IBD patients. 22 In detail, CD patients’ intestinal mucosa was enriched in RNAs belonging to Hepeviridae, a family of RNA eukaryotic viruses normally causing hepatitis in mammals, whereas UC patients displayed Hepadnaviridae transcripts (hepatitis B virus (HBV) belongs to this DNA eukaryotic viral family). These results pointed out an association of these eukaryotic viral families with IBD etiopathogenesis and further studies are urgently needed to demonstrate their role in causing chronic intestinal inflammation. An approach to successfully associate viral triggers with IBD etiopathogenesis might be the metagenome profiling of first-degree relatives of IBD patients that do not have IBD yet. The first-degree relative follow-up may allow the tracking of any IBD-related symptom manifestation. When some subjects develop the disease, their metagenome signatures can be associated with IBD, ultimately identifying potential viral triggers. A similar study has been already initiated in the GEM project (https://crohnsandcolitis.ca/Research/Funded-research/The-gem-project).
Viruses as early triggers for a disease have been already explored in previous studies and clinical evidence is already available for tumours. 81 In fact, HBV, HCV, human papillomavirus, human herpesvirus 8, Merkel cell polyomavirus and HTLV-1 are responsible for the 80% of hepatocellular carcinomas. In all these cases, however, viral infections occur in the tissues in which cancer develops, because their opportunistic lifecycle leads to uncontrolled cell proliferation and finally to tumorigenesis.
In contrast, non-enterotropic eukaryotic viral entities may infect mucosa, apparently without symptomatic manifestation, but are likely to confer a virotype 76 to the host that might latently stimulate its mucosal immune response that, in turn, initiates IBD pathogenesis. To the best of our knowledge, neither scientific nor clinical demonstrations confirming these novel and exceptional insights for IBD aetiogenesis are available. Once the viral species infecting patients in the early phase of intestinal inflammation are identified and characterized, it will be possible to identify early biomarkers predictive of the disease and engineer specific siRNAs or antiviral drugs to treat IBD patients, thus providing a real breakthrough in the field.
Perspective
Researchers and clinicians have always made great efforts to define the correct microbiota composition characterizing its pathogenesis. Nevertheless, scientific evidence stating that alterations in microbiota are the real cause of the initiation is yet to be established. Many efforts are being made with the unique purpose of defining the actor(s) in this scenario. Cutting-edge NGS technologies, -omics approaches and computational biology are the ultimate helpers for clarifying this, because they may build in silico the complex structure of the gut microbiota (Figure 2). By performing NGS and -omics at a single-cell resolution we will possibly identify which cell type(s) represent the primary targets of the gut opportunistic colonizers and how they affect host cells’ transcriptomics, metabolomics and immunity, eventually pointing out novel molecular mechanisms underlying IBD etiopathogenesis. The results will be useful for the development of novel and effective therapies,
82
not merely treating symptoms, but counteracting the primary cause of chronic intestinal inflammation.
The multi-omic approach. Exploiting the computational biology, it will be possible to combine all -omic results and provide a composite picture of inflammatory bowel disease (IBD) etiopathogenesis.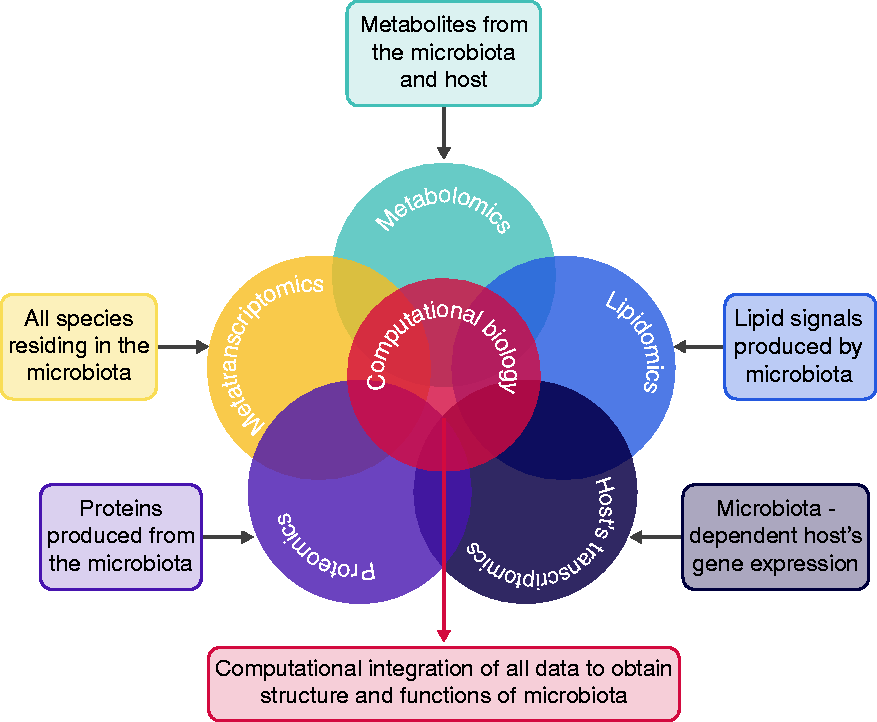
Footnotes
Declaration of conflicting interests
The author(s) declare that there is no conflict of interest.
Ethics approval
For animal studies mentioned in this review, please refer to the original articles.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Informed consent
For informed consent required for human studies mentioned in this review, please refer to the original articles.