
Abstract
Background
The mechanisms of macrophages/monocytes in autoimmune hepatitis (AIH) remain unclear. We investigated the role of receptor-interacting protein kinase 3 (RIP3), a key inflammatory signal adapter, in macrophage/monocyte activation in AIH.
Methods
Liver tissues and monocytes from patients were collected to evaluate the relationship between macrophage activation and RIP3 by double-immunofluorescence and Western blotting. RAW264.7 macrophages were used to study the regulation of RIP3 signaling on inflammatory cytokines.
Results
Compared to the hepatic cyst, the majority of accumulated macrophages expressed RIP3 in AIH liver tissues. Moreover, RIP3 expression of monocytes was correlated with the levels of serum hepatic enzyme in AIH. Furthermore, RIP3 signaling was activated by lipopolysaccharide in RAW264.7 macrophages, which was accompanied with upregulated interleukin (IL)-1β, IL-6, and IL-10 and downregulated IL-4 and transforming growth factor-β. Notably, necrostatin-1, the specific inhibitor of the RIP3 signaling pathway, and 6-thioguanine (6-TG), the active metabolite of azathioprine, predominantly reduced IL-6 production compared to other cytokines. Moreover, the gene level of IL-6 was dramatically increased in AIH liver tissues.
Conclusions
RIP3 signaling is involved in macrophage/monocyte activation in AIH and mediates IL-6 production, and is a novel molecular mechanism of 6-TG, indicating that it might be a promising therapeutic target for AIH treatment.
Key summary
Our study determined a novel molecular mechanism of macrophage/monocyte activation and inflammatory response in autoimmune hepatitis (AIH). Moreover, the role of receptor-interacting protein kinase 3 (RIP3) in human AIH disease was reported for the first time. In addition, this study points out the relationship between interleukin (IL)-6 and AIH, and IL-6 can be downregulated through inhibiting the RIP3 signaling of macrophages. Finally, our investigation provided a novel mechanism for the effect of thiopurines on the immune system.
Introduction
Autoimmune hepatitis (AIH) is a progressive autoimmune-mediated inflammatory disease of the liver. 1 Macrophages play a central role in maintaining immune homeostasis as well as in initiating and controlling inflammatory responses in the liver.2,3 Persistent and excessive activation of macrophages results in pathological inflammation and fibrosis in chronic liver diseases.4–7 Previous research suggests that activated macrophages are present in portal infiltrates and at sites of interface hepatitis in AIH.8–11 Nonetheless, until now little has been known about the molecular mechanisms of macrophage activation in AIH.
Receptor-interacting protein kinase 3 (RIP3) has been increasingly recognized as a key inflammatory signal adapter, which mediates inflammation through necroptosis as well as non-necroptosis function.12–14 Evidence of RIP3-deficient mice exhibiting reduced inflammation in many inflammatory disease models strongly confirmed the relevance of RIP3 signaling with inflammatory response.15–17 Deutsch et al. reported that blockading RIP3 ameliorated Concanavalin A-induced hepatitis. 18 However, the premise mechanism still remains elusive and the role of RIP3 in human AIH disease has not yet been reported. As mentioned above, activated macrophages play a crucial role in the development of AIH, therefore we speculated that RIP3 might be involved in the macrophage activation and inflammatory response in AIH.
The primary function of macrophages involves cytokine production, 19 which is essential for initiating inflammatory response and forms a highly complex network for immune system homeostasis. 20 The dysregulation of inflammatory cytokines leads to the breaking of immune microenvironment balance, shifting the local environment toward a proinflammatory state and resulting in tissue damage. 21 Hence, understanding the effect of RIP3 on inflammatory cytokines is crucial in clarifying the pathogenesis of AIH.
Here, we investigated the role of RIP3 in macrophage activation in AIH liver tissues, and confirmed the relationship between RIP3 activity and the severity of liver injury. Further, in order to gain insight into the development of AIH, we explored the regulation of RIP3 signaling on inflammatory cytokines.
Materials and methods
Human liver biopsies
Liver biopsy specimens were collected from seven patients with AIH (age 55.6 ± 16.1 years), and four patients with hepatic cyst (age 54.8 ± 6.2 years), who were enrolled in this study from the Department of Gastroenterology and Hepatology in Tianjin Medical University General Hospital. The diagnosis of AIH was performed according to simplified criteria for the diagnosis of autoimmune hepatitis. 22 Biopsies were immediately snap-frozen in liquid nitrogen and stored at –80℃ for immunofluorescence analysis. The clinical and pathological data of patients are documented in Supplementary Table S1. All patients gave informed consent to the study, which was performed in accordance with the ethical guidelines of the 1975 Declaration of Helsinki and approved by the Tianjin Medical University Ethics Committee (January 2015).
Human CD14+ monocytes
CD14+ monocytes were isolated from 11 patients with AIH and four healthy volunteers. Blood samples of AIH patients and healthy volunteers were collected to separate peripheral blood mononuclear cells (PBMCs) by Ficoll-Hypaque density-gradient centrifugation. PBMCs were further purified by the usage of CD14 microbeads (Miltenyi Biotec, Germany) according to the manufacturer’s protocol. Clinical characteristics of AIH patients and healthy volunteers are presented in Supplementary Table S2.
Immunofluorescence
Double immunofluorescence analyses for CD68/RIP3 were performed with 4-µm-thick frozen sections. Slides were fixed with acetone and blocked with 5% bovine serum albumin, then incubated with antibodies against CD68 (Abcam, UK) and RIP3 (Abcam, UK), and further incubated with Alexa Fluor 488 (Abcam, UK) and Alexa Fluor 568 antibody (Abcam, UK). Nuclear staining was achieved by 4’, 6-diamidino-2-phenylindol (DAPI).
Cell culture
RAW264.7 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA), 100 U/ml penicillin, 100 mg/ml streptomycin and incubated at 37℃ at an atmosphere of 5% CO2.
Western blot analysis
CD14+ monocytes and RAW264.7 cells were lysed with radioimmunoprecipitation assay lysis buffer (Solarbio, China). Whole extracts were prepared, and protein concentration was detected using a bicinchoninic acid protein assay kit (Solarbio, China). A total of 20 µg of total proteins were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins were transferred in polyvinylidene difluoride membrane (Bio-Rad Laboratories, USA) and blocked with Tris-buffered saline-Tween (TBS-T) containing 5% non-fat dry milk. Anti-RIP1 (Abcam, UK), anti-RIP3 (Abcam, UK), anti-mixed lineage kinase domain-like (MLKL) (Abcam, UK), and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Cell Signaling Technology, USA) antibodies were diluted in TBS-T containing 3% non-fat dry milk and incubated overnight at 4℃. Membranes were washed in TBS-T, incubated for one hour with horseradish peroxidase-conjugated secondary antibody. Proteins were visualized with an enhanced chemiluminescence kit (Thermo Scientific, USA). Densitometric analyses of the blots were performed using the software ImageQuant. Subsequently, the RIP3 expression levels of monocytes were semi-quantified and ascribed a hierarchical score as (–) designating low level of RIP3, relative density < 0.3, (+) designating mild level of RIP3, relative density > 0.3, but < 1.0, (++) designating high level of RIP3, relative density > 1.0. All experiments were repeated three times.
Quantitative real-time polymerase chain reaction (PCR)
Total RNA was extracted from RAW264.7 cells and liver tissues using TRIzol reagents (Gibco, USA), and the first-strand complementary DNA was synthesized using Thermoscript reverse transcription (RT)-PCR synthesis kit (Fermentas, USA) according to the manufacturer’s instructions. Real-time quantitative PCR analyses for messenger RNA (mRNA) of tumor necrosis factor alpha (TNF-α), IL-1β, IL-6, IL-10, IL-4, transforming growth factor-beta (TGF-β) and GAPDH were performed by using Thermoscript RT-qPCR kits (Fermentas, USA) in an ABI Prizm step-one plus real-time PCR System (Applied Biosystems, USA). The mRNA level of GAPDH was used as an internal control. Relative expression levels were calculated according to the standard 2−ΔΔCt method. All experiments were performed in triplicate and repeated at least three times.
Statistical analysis
Data are presented as means ± standard deviation (SD) and were analyzed using SPSS 17.0 software. Student’s t test was performed for normally distributed continuous variables. Kruskal–Wallis test followed by Mann–Whitney U test was performed for non-normally distributed continuous variables. In all cases, values of p < 0.05 were considered to be statistically significant. Error bars represent the SD.
Results
Accumulated macrophages express RIP3 in AIH liver tissues
In comparison to the hepatic cyst (n = 4), extensive accumulation of CD68+ macrophages were detected within AIH liver tissues (n = 7) (4.75 ± 0.96 vs 28.86 ± 6.23, p < 0.05). Furthermore, the majority of CD68+ macrophages were co-localized with RIP3, an important inflammatory signal adapter, in AIH liver tissues. In contrast, the co-localization of CD68+ macrophages and RIP3 was rarely observed in the liver tissues of hepatic cyst (16.4 ± 5.7 vs 0.5 ± 0.6, p < 0.05). These findings indicated that RIP3 is associated with macrophage activation and might be involved in the inflammatory response of AIH (Figure 1).
Accumulated macrophages express RIP3 in AIH liver tissues. Double-immunofluorescence staining for CD68 and RIP3 in liver tissues of patients with AIH (n = 7) and hepatic cyst (n = 4). Over-accumulated CD68+ macrophages (green) and elevated RIP3 staining (red) were observed in AIH frozen liver tissues ((a) and (c)), while CD68+ macrophages and RIP3 staining were quite rare in liver tissues of hepatic cyst ((b) and (d)). In AIH liver tissues, the majority of CD68+ macrophages were co-localized with RIP3 (g), whereas rare CD68+ macrophages were co-localized with RIP3 in liver tissues of the hepatic cyst (h) (scale bar = 100 µm). RIP3: receptor-interacting protein kinase 3; AIH: autoimmune hepatitis.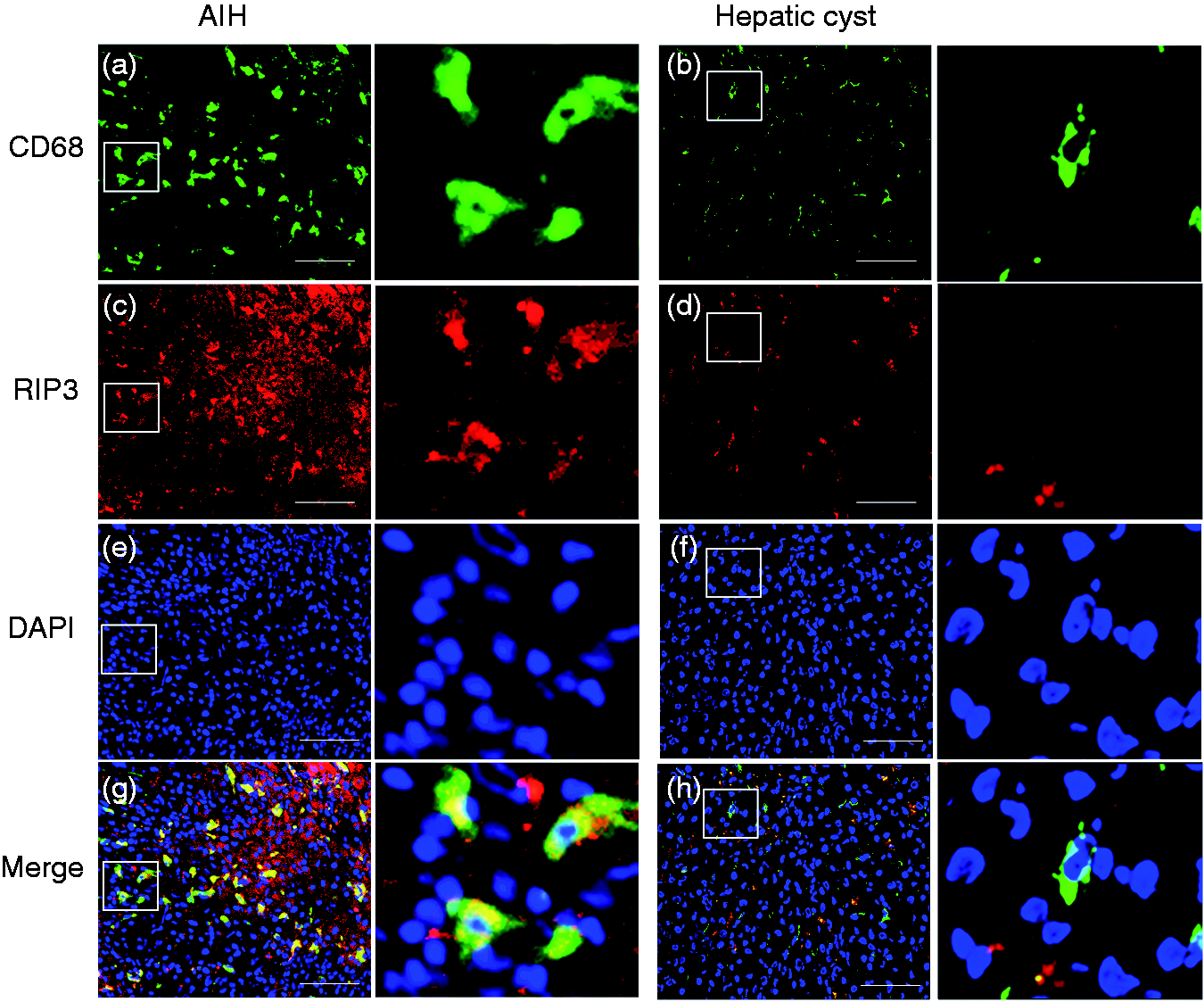
RIP3 activities of monocytes are associated with the severity of liver injury
Next, we analyzed the relationship of RIP3 signaling and liver inflammation in AIH. RIP3 expression of peripheral blood monocytes from age- and gender-matched AIH patients (n = 11) and healthy controls (n = 4) was examined by Western blot and semi-quantified according to the density of blots. The serum levels of alanine aminotransferase (ALT) were significantly higher in AIH patients, whose monocytes expressed high levels of RIP3. In comparison, the serum levels of ALT were lower in AIH patients and healthy patients, whose monocytes expressed milder or lower levels of RIP3. A similar correlation was observed between serum levels of aspartate aminotransferase (AST), glutamyl transpeptidase (GGT) and RIP3 expression of monocytes in AIH patients and controls. However, owing to the limited sample size, no significant association was observed between serum levels of immunoglobulin G (IgG), antinuclear antibody titers and RIP3 expression of monocytes (Figure 2(b)). These findings suggested that RIP3 activities of monocytes are correlated with the extent of liver inflammation in AIH.
RIP3 activities of monocytes are associated with the severity of liver injury. (a) RIP3 expression of monocytes from AIH patients (n = 11) and healthy controls (n = 4) was examined by Western blot. Representative images are shown. (b). RIP3 expression was semi-quantified and ascribed a hierarchical score according to the density of blots. The serum levels of ALT, AST, ALP, GGT, IgG, and ANA titers of patients with different RIP3 scores were analyzed by Mann–Whitney U test. (c) RIP3 expression of monocytes was notably reduced in patients (n = 3) after treatment, which coincided with a significant reduction in the serum ALT and AST levels. (d) RIP3 expression levels remained unchanged in patients (n = 3) without significant reduction of ALT and AST levels. *p < 0.05, **p < 0.005, ***p < 0.0005. AIH: autoimmune hepatitis; ALT: alanine aminotransferase; ALP: alkaline phosphatase; ANA: antinuclear antibodies; AST: aspartate aminotransferase; GGT: glutamyl transpeptidase; IgG: immunoglobulin G; n.s.: not significant; RIP3: receptor-interacting protein kinase.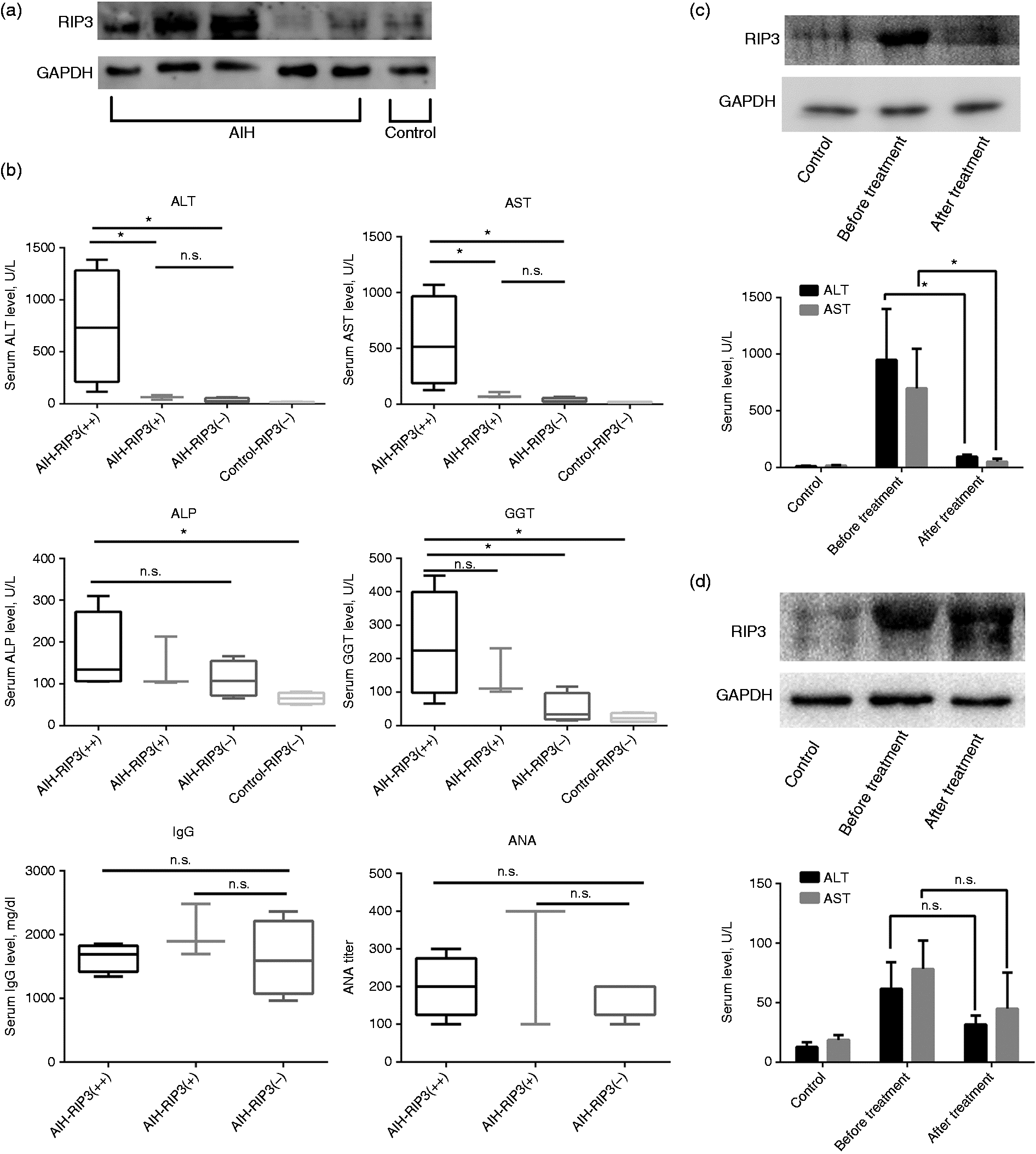
Furthermore, RIP3 expression of monocytes was compared in patients prior to and following treatment (n = 6). RIP3 expression of monocytes was notably reduced in patients (n = 3) after treatment, which coincided with a significant reduction of the serum ALT and AST levels (Figure 2(c)), whereas RIP3 expression levels remained unchanged in patients (n = 3) without a significant reduction of ALT and AST levels (Figure 2(d)). These data synergistically confirmed the relationship between RIP3 activities of monocytes and liver inflammation in AIH.
Activated macrophages express upregulated RIP3 signaling
To further confirm the link between RIP3 signaling and the activation of macrophages, RAW264.7 macrophages were stimulated by lipopolysaccharide (LPS) in vitro. Macrophages were exposed to varying concentrations of LPS (0 µg/ml, 1 µg/ml, 3 µg/ml, and 10 µg/ml) for 24 hours. The expression levels of RIP1 (the major upstream activator of RIP3), RIP3, and MLKL (the direct downstream effector of RIP3) were dose-dependently upregulated following LPS stimulation (Figure 3(a)), indicating that RIP3 signaling is activated during the activation of macrophages.
Activated macrophages express upregulated RIP3 signaling. (a) RAW264.7 macrophages were exposed to varying concentrations of LPS (0 µg/ml, 1 µg/ml, 3 µg/ml, 6 µg/ml and 10 µg/ml) for 24 hours. Expression levels of RIP1, RIP3, and MLKL were examined by Western blot. RIP1, RIP3, and MLKL levels were dose-dependently upregulated following LPS stimulation. (b) RAW264.7 macrophages were incubated with LPS (3 µg/ml) and/or Nec-1 (100 µM) as described in images. Western blotting analysis showed that RIP1, RIP3, and MLKL levels were downregulated in macrophages after co-treatment with Nec-1 compared to LPS alone treatment. *p < 0.05, **p < 0.005, ***p < 0.0005. LPS: lipopolysaccharide; MLKL: mixed lineage kinase domain-like; Nec-1: necrostatin-1; RIP: receptor-interacting protein kinase.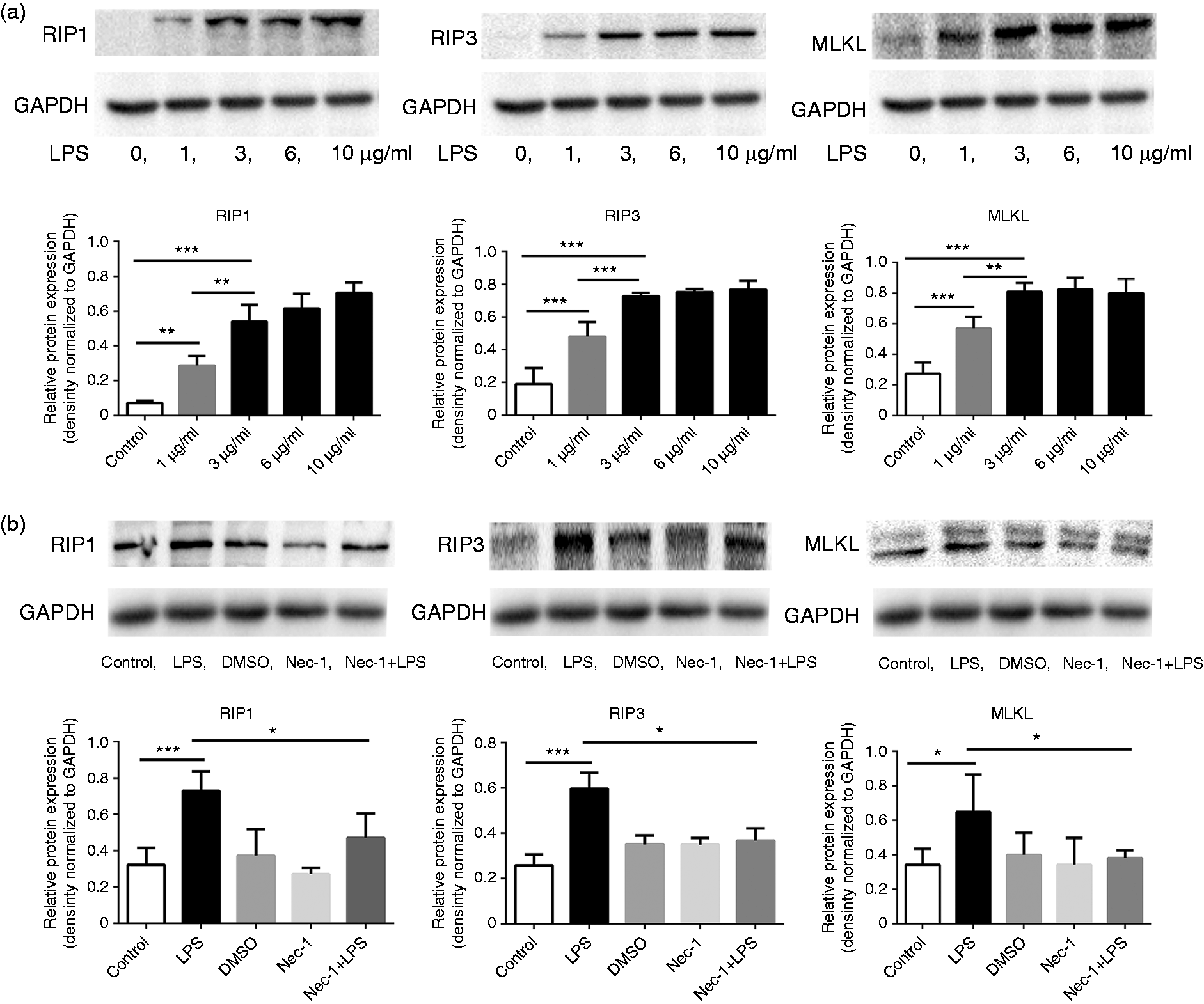
Necrostatin-1 (Nec-1) is a specific inhibitor of the RIP3 signaling pathway. Figure 3(b) shows that Nec-1 consistently downregulated the expression of RIP1, RIP3, and MLKL in LPS-stimulated RAW264.7 macrophages.
RIP3 activation regulates IL-6 production in macrophages
To further explore the mechanism of RIP3 activation in macrophages leading to liver inflammation, we analyzed the regulation of RIP3 signaling in an inflammatory cytokine microenvironment. Upon activation of the RIP3 signaling by LPS in RAW264.7 macrophages, the mRNA expression of IL-1β, IL-6, and IL-10 was upregulated, whereas the expression of IL-4 and TGF-β was downregulated. Interestingly, Nec-1, which inhibits the RIP3 signaling pathway, downregulated the IL-1β, IL-6 and IL-10 expression in LPS-stimulated RAW264.7 macrophages, and upregulated the expression of IL-4 and TGF-β. Of note, the change of IL-6 was most notable compared to other cytokines (IL-6: 6.10-fold, IL-1β: 1.75-fold, IL-10: 2.00-fold, IL-4: 2.23-fold, TGF-β 2.10-fold) (Figure 4(a)). There was no significant change in TNF-α (data not shown). These findings suggest that RIP3 signaling was involved in the regulation of macrophage-related cytokines, in particular IL-6.
RIP3 activation regulates IL-6 production in macrophages. (a) RAW264.7 macrophages were incubated with LPS (3 µg/ml) and/or Nec-1 (100 µM). The levels of IL-1β, IL-6, IL-10, IL-4 and TGF-β in macrophages of each group were analyzed by RT-PCR. (b) The IL-1β, IL-6, IL-10, IL-4 and TGF-β levels of liver tissue from patients with AIH and hepatic cyst were analyzed by RT-PCR. *p < 0.05, **p < 0.005, ***p < 0.0005. AIH: autoimmune hepatitis; IL: interleukin; LPS: lipopolysaccharide; MLKL: mixed lineage kinase domain-like; Nec-1: necrostatin-1; n.s.: not significant; RIP: receptor-interacting protein kinase; TGF-β: transforming growth factor-beta; RT-PCR: reverse transcription-polymerase chain reaction.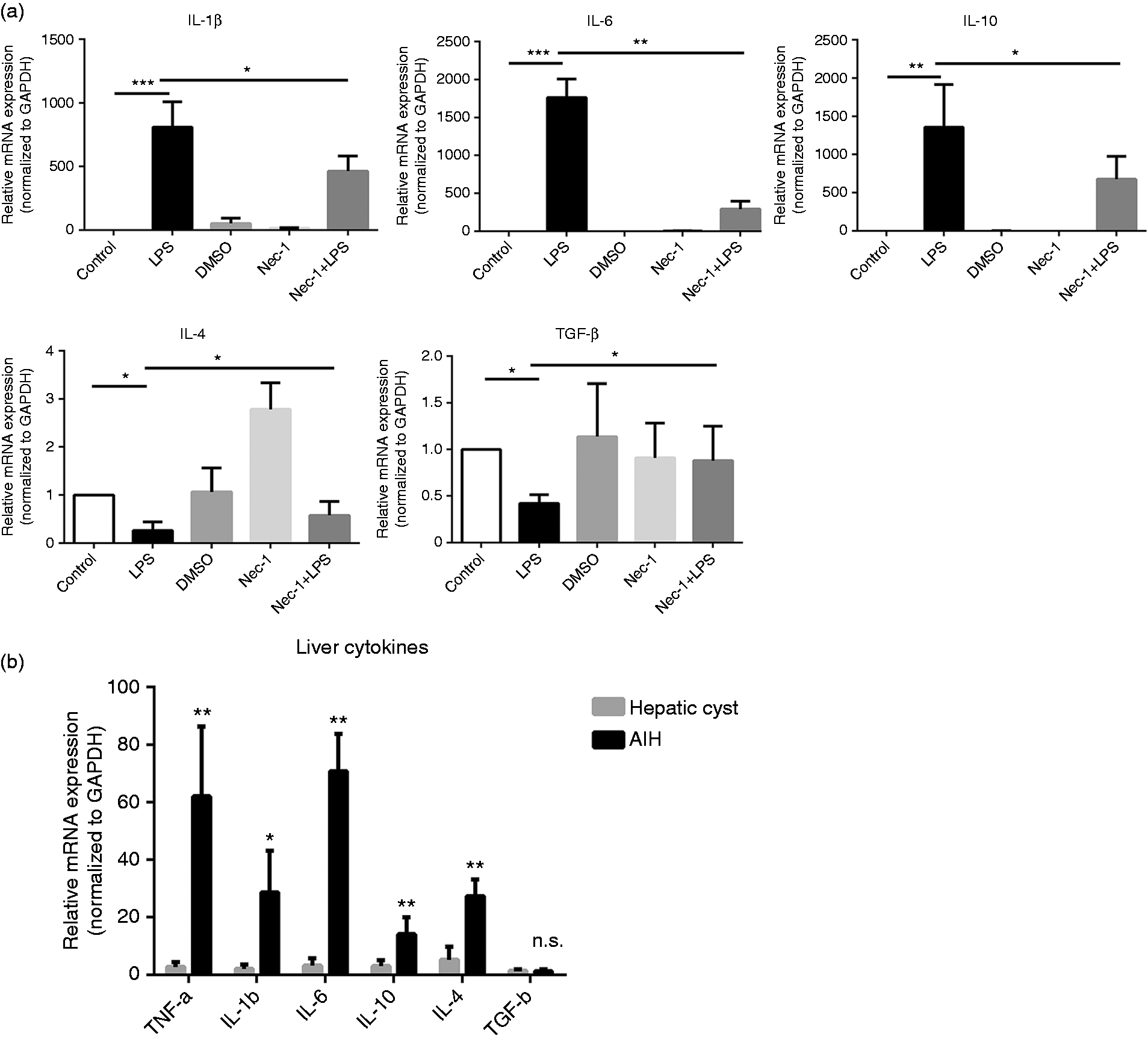
Importantly, compared to hepatic cyst (n = 4), the mRNA expression of IL-6 and TNF-α was dramatically increased in AIH liver tissues (n = 7) compared with other cytokines (IL-6: 21.28-fold, TNF-α: 22.20-fold, IL-1β: 13.24-fold, IL-10: 4.53-fold, IL-4: 5.13-fold). There was no significant change in TGF-β (data not shown) (Figure 4(b)).
Immunosuppressant 6-thioguanine (6-TG) inhibits RIP3 signaling of macrophages
Azathioprine is the first-line immunosuppressive drug for the treatment of AIH. To determine whether RIP3 signaling is targeted by azathioprine, RAW264.7 macrophages were incubated with LPS (3 µg/ml) and/or 6-TG (10 µM), the active metabolite of azathioprine. Upon LPS stimulation, 6-TG significantly downregulated the protein expression of RIP1, RIP3, and MLKL (Figure 5(a)). Also consistent with the effect of Nec-1, 6-TG decreased the mRNA expression of IL-1β, IL-6, and IL-10 and increased IL-4 and TGF-β expression in LPS-stimulated RAW264.7 (Figure 5(b)).
Immunosuppressant 6-TG inhibits the RIP3 signaling of macrophages. RAW264.7 macrophages were treated with LPS (3 µg/ml) and/or 6-TG (10 µM) as described in images. (a) Western blotting analysis showed that co-treatment with 6-TG downregulated the expression levels of RIP1, RIP3, and MLKL, which were upregulated by LPS. (b) Effect of 6-TG on the cytokine production of macrophages. *p < 0.05, **p < 0.005, ***p < 0.0005. RIP: receptor-interacting protein kinase; LPS: lipopolysaccharide; 6-TG: 6-thioguanine; MLKL: mixed lineage kinase domain-like.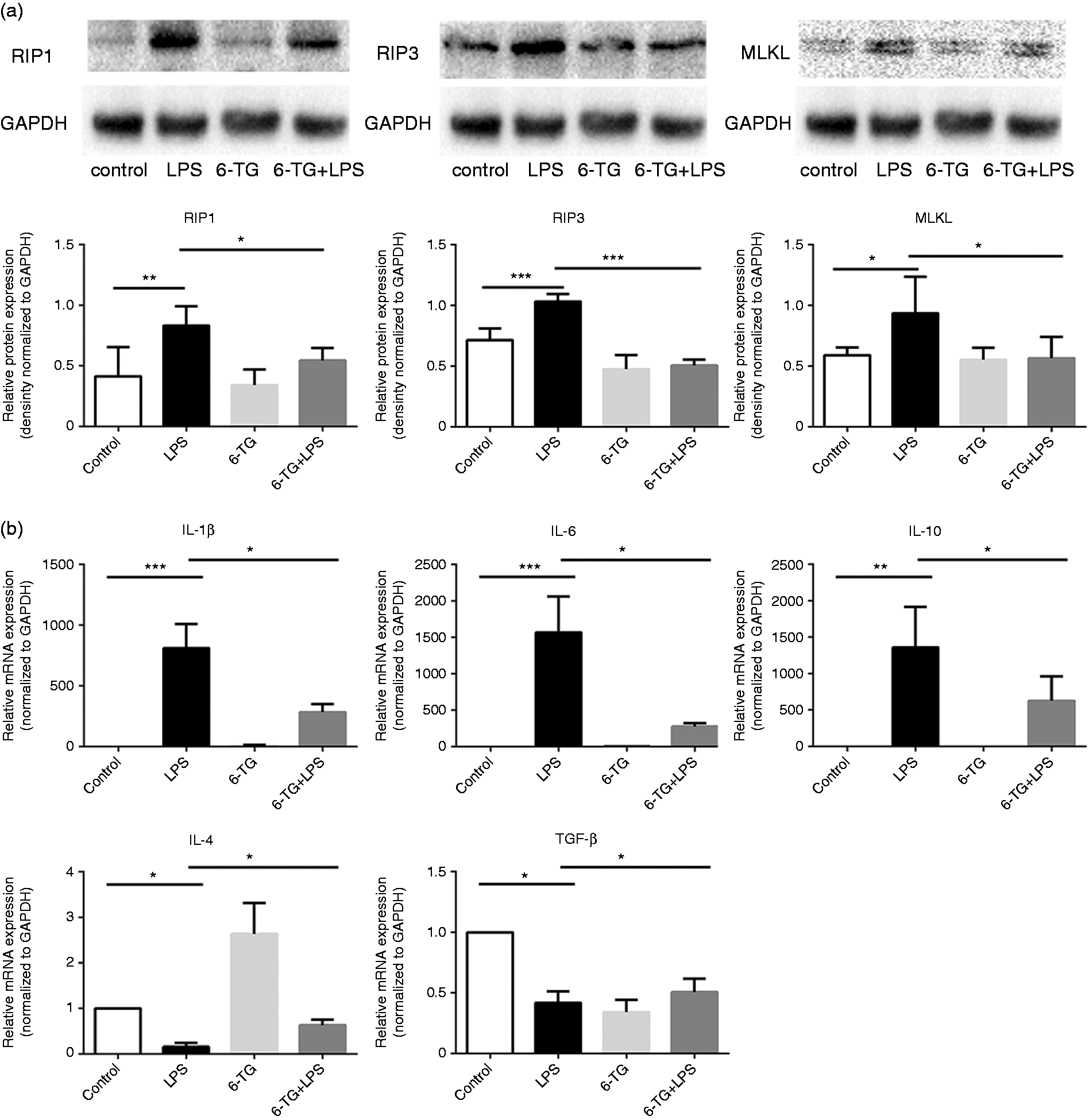
Discussion
In the present study, RIP3, an inflammatory signal adapter, is highly correlated with macrophage/monocyte activation and serum ALT/AST levels in patients with AIH, indicating that the RIP3 signal plays important roles in macrophage/monocyte activities and subsequently liver inflammation of AIH. Furthermore, RIP3 predominantly regulated IL-6 production of RAW264.7 macrophages, which was in line with the cytokine profiles of AIH liver tissues. Moreover, RIP3 signaling is demonstrated as a novel molecular mechanism of 6-TG. These results collectively provide important insights into the cellular and molecular mechanism of liver inflammation, which may be the potential target for AIH therapy.
Macrophages are implicated in the pathological inflammation and fibrosis of liver tissues.4–7 Previous studies illustrated that activated macrophages are present in portal infiltrates of AIH.8–11 Our study reported activated macrophages highly expressed RIP3 in AIH liver tissues, revealing a possible molecular mechanism for macrophages activation, which is involved in the liver inflammation of AIH. Moreover, the expression levels of RIP3 in monocytes were correlated with serum ALT/AST levels, which are important clinical parameters for hepatocyte injury and disease activity of AIH. These findings confirmed that RIP3 activities of monocytes are involved in the liver inflammation of AIH and may be a potential biomarker for following AIH disease activity. Importantly, RIP3 expression of monocytes directly reflects the status of immune cells, presenting a novel immune-based biomarker.
Our investigation noted that RIP3 regulated IL-6 production, which was dramatically increased in AIH liver tissues. IL-6 has been demonstrated to be a B-cell-stimulating factor that drives IgG production 23 and is a potent factor in switching the induction of regulatory T cell (Treg) to pathogenic IL-17-producing T-helper cells (Th17) in other autoimmune diseases.24,25 Importantly, the anti-IL-6 receptor antibody tocilizumab has been shown to be effective in several autoimmune diseases such as uveitis, systemic lupus erythematosus, and Crohn’s disease.26–28 Consistent with other autoimmune diseases, IgG and Th17/Treg imbalance are of pathological importance for the development of AIH. 29 However, to date, there is limited research into the role of IL-6 in AIH. The data obtained from our study indicate that IL-6 plays an important part in the development of AIH and is a possible molecular mechanism in the RIP3 activation of macrophages leading to the progression of AIH.
When compared with Nec-1, 6-TG performs similar effects on RIP3 signaling. Although inhibition of purine nucleotide biosynthesis based on random incorporation of 6-TG nucleotides into DNA has been suggested as a therapeutic mechanism, 30 a general inhibition of nucleic acid synthesis is not sufficient to explain the specific effects of thiopurines on the immune system.31,32 Our investigation showed that 6-TG inhibited the RIP3 signaling of macrophages, providing a novel molecular mechanism for the effect of thiopurines on the immune system. On the other hand, RIP3 signaling was targeted by immunosuppressant 6-TG, indicating that RIP3 could be the therapeutic target of AIH.
In conclusion, the inflammatory signal adapter RIP3 is a novel endogenous molecular factor involved in macrophage/monocyte activation in AIH and regulates IL-6 production, which plays a pathological role in AIH. Therefore, regulating RIP3 signaling of macrophages along with tightly controlling IL-6 production could perhaps be a promising strategy in the management of AIH inflammatory response and progress.
Footnotes
Acknowledgments
We thank Zhen Wang and Fang Zhang for their support of some experimental methods.
Declaration of conflicting interests
None declared.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81200282 and No. 81470834).
Ethics approval
This study was performed in accordance with the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Tianjin Medical University Ethics Committee (January 2015).
Informed consent
All patients gave informed consent to this study.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.