
Abstract
Background
Oesophageal candidiasis is a common, usually self-limiting opportunistic infection, but long-term infection with Candida is known to predispose to oral and oesophageal squamous cell cancer (SCC). Permissive factors that lead to immune deficiencies can underlie persistent or recurring candidiasis, called chronic mucocutaneous candidiasis (CMC). Secondary immune deficiencies are most often due to human immunodeficiency virus (HIV) infection, antibiotic use and immunosuppressive treatment (steroids, chemotherapy). Inborn errors of the immune system (primary immune deficiencies) can present with isolated CMC known as CMC disease (CMCD), which is most often found in patients with autoimmune polyendocrinopathy syndrome type 1 (APS1)/APECED or in patients with an underlying gain-of-function STAT1 mutation (GOF-STAT1).
Objective
To describe a new form of inherited/familial CMC with a high risk for developing squamous cell carcinoma of the oesophagus, due to a gain-of-function mutation in the STAT1 gene.
Methods and results
This report describes a family of patients with CMC with confirmed GOF-STAT1 mutation. These patients usually present with CMCD in childhood, have severe oral and oesophageal candidiasis accompanied by severe difficulty swallowing, chest pain, heartburn, and are at risk of developing oral and/or oesophageal SCC. This case series describes six patients in three generations of the same family, two of whom developed and died of SCC. We recommend regular endoscopic surveillance to detect early oesophageal neoplasia in patients with CMCD as well as urgent endoscopy in symptomatic patients.
Conclusion
CMC is not a well-recognised condition in gastroenterology practice and clinicians need to be aware of the genetics of the condition as well as the risk for oesophageal cancer so that they can counsel their patients and arrange surveillance appropriately.
Keywords
Introduction
Candida is an opportunistic yeast, colonising gastrointestinal and urogenital mucosa in about 30–50% of healthy humans without causing infection. Candida overgrowth requires permissive circumstances which occur when the immune system is damaged, leading to either primary or secondary immune deficiencies, which, depending on the underlying immune defect, will lead to infections of the skin, nails and mucosa (oral, oesophageal, genital) with Candida, coined chronic mucocutaneous candidiasis (CMC), or to invasive disease (sepsis) with internal organ involvement. Secondary CMC can be precipitated by a range of factors, the most common being secondary immune deficiency caused by human immunodeficiency virus (HIV) infection, when CD4 T lymphocyte counts fall to <100 cells/μl, precipitating acquired immune deficiency syndrome (AIDS). Other frequent precipitating factors include use of antibiotics and immunosuppressive drugs such as long-term systemic or inhaled corticosteroids, chemotherapy for malignancies, as well as other diseases such as diabetes or local factors such as dentures. 1
In contrast to the above, in patients with inborn (genetic) errors of the immune system known as primary immune deficiencies (PIDs), CMC can present as a syndrome with chronic, persistent or recurrent, debilitating Candida infection of the skin, nails and mucous membranes without an obvious cause as defined above (see Figures 2 and 3) In these patients, CMC, especially oral Candida infection (thrush), may occur either in isolation (CMC disease – CMCD), as part of well-defined PID syndromes or part of a broad, severe immune deficiency. Patients with the autosomal dominant (AD) hyper IgE syndrome (HIES) suffer with CMC as well as severe skin and pulmonary staphylococcal infections. This is caused by a mutation in the signal transducer and activator of transcription 3 (STAT3) gene resulting in a faulty STAT3 function, leading to significantly reduced levels of CD4 T helper (Th)-17 cells and circulating interleukin (IL)-17 and IL-22 cytokines. CMC can also be part of a complex clinical phenotype caused by severe immune deficiencies affecting T lymphocytes, where patients suffer with susceptibility to a broad range of microorganisms rather than selective fungal infections. Accumulating evidence suggests that mucocutaneous fungal infections accompany a variety of defects that disrupt the Th-17 pathway, 2 while protection against invasive disease is mediated by phagocytes, and indeed CMC patients rarely if ever develop invasive fungal disease. 1
Oesophageal candidiasis can present as dysphagia, odynophagia, retrosternal chest pain, or can be an incidental finding at endoscopy. Its typical endoscopic appearance is of white plaques which persist despite water flushes. Oesophageal brushings and biopsy show the characteristic appearance of yeasts and pseudo hyphae. The most common organism associated with this is Candida albicans, although other strains such as C. glabarata and C. tropicalis are increasingly being isolated. Common endoscopic findings associated with oesophageal candidiasis include oesophageal ulcer, gastric ulcer and severe atrophic gastritis. 3 Rare complications include ulceration, haemorrhage or oesophageal obstruction from stricture or fistulation to bronchial tree. 4 For most patients one course of antifungal treatment is sufficient to resolve the problem, and repeat gastroscopy is very rarely needed.
In rare patients with an underlying PID, the oesophageal candidiasis presents as CMCD with recurrent or persistent thrush despite multiple courses of antifungal treatment; it is usually accompanied by candidiasis of the skin, nails and other mucous membranes, and often presents in early childhood and frequently involves multiple family members.
CMCD is known to be caused by at least five different mutations affecting the IL-17 pathways, 2 but the two most common causes of CMCD are autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED), also known as autoimmune polyendocrinopathy syndrome type 1 (APS1), and AD gain-of-function (GOF) gene mutation of the signalling protein STAT1 (signal transducer and activator of transcription 1).
In APECED/APS1, CMC is seen in >95% of patients, often as the first presenting symptom of disease, followed by the development of a triad of hypoparathyroidism, adrenal and gonadal failure, as well as other endocrine organ involvement (e.g. diabetes). APECED is caused by an inborn error (mutation) of the AIRE gene, inherited in an autosomal recessive manner. AIRE is a transcription factor that promotes the ectopic presentation of self-antigens in the thymic medulla, ensuring removal of self-reactive T lymphocytes in the process of thymic T cell maturation. Defective AIRE function and failed removal of auto-reactive T cells results in autoimmunity, causing endocrine organ failure; however, why these patients develop CMC was a mystery until we and others demonstrated in APECED/APS1 patients the production of neutralising autoantibodies to Th-17 cytokines IL-17A-F and IL-22,5,6 disrupting Th-17 mediated fungal immunity, which explained why these patients develop CMC in the context of the underlying autoimmunity.
In the other largest group of patients with CMCD, we and others recently identified a GOF mutation in the STAT1 gene as the underlying cause.7,8 Although the pathogenic mechanisms are still not fully understood, it is thought that the genetic defect results in exaggerated Th1 responses which down regulate STAT3/Th17 immunity, leading to reduced production of IL-17 and IL-22.
9
In these patients, there have been sporadic reports of a link between CMC and oesophageal squamous cell carcinoma,
10
confirmed in a recent international review of these rare patients.
11
In view of the increasingly recognised association of SCC and GOF-STAT1-CMC, we present a case series in a family of three generations with this disease (Figure 1).
Family tree of CMC patients. Appearance of the nails in CMC. Appearance of the tongue in CMC.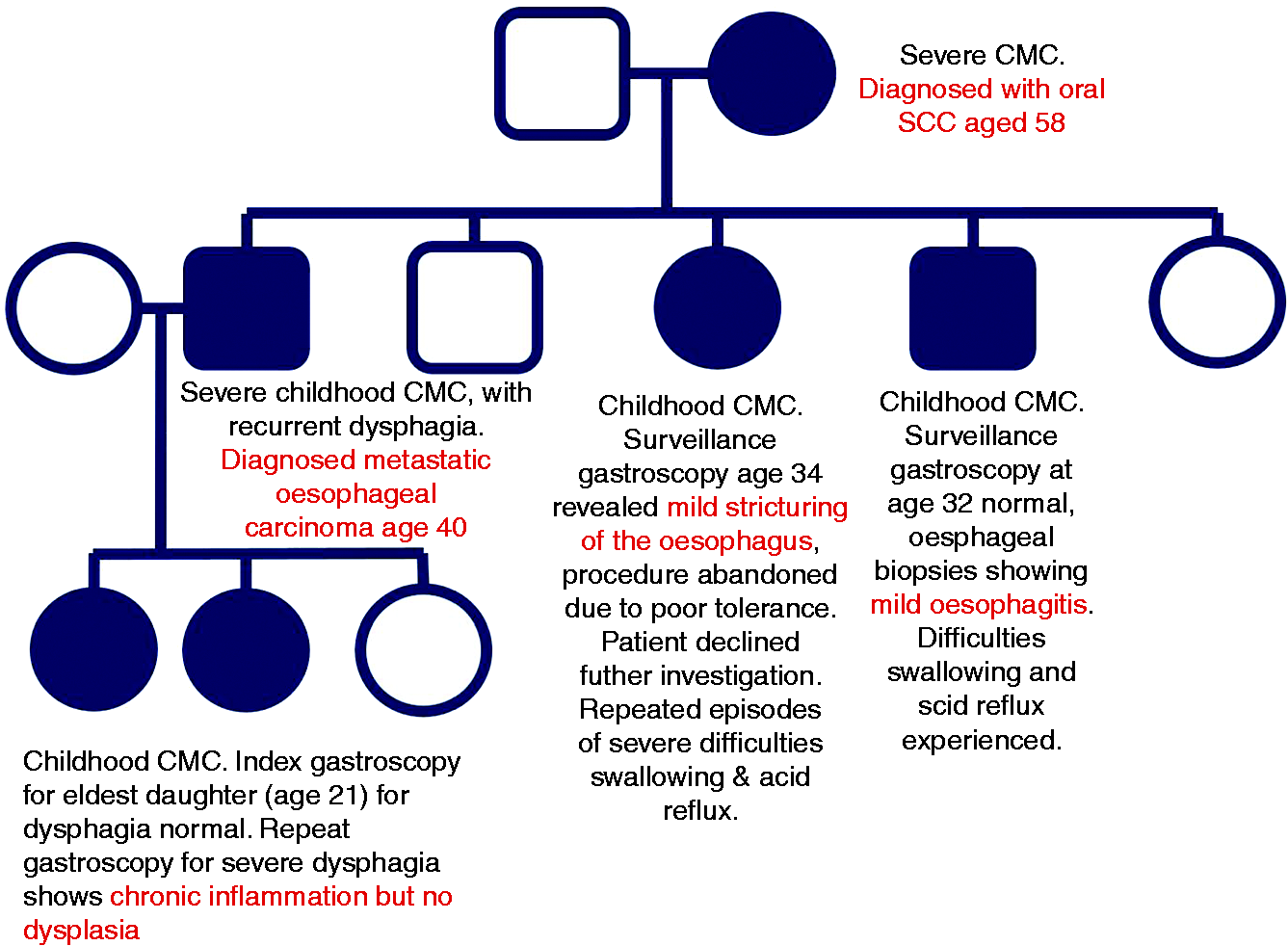
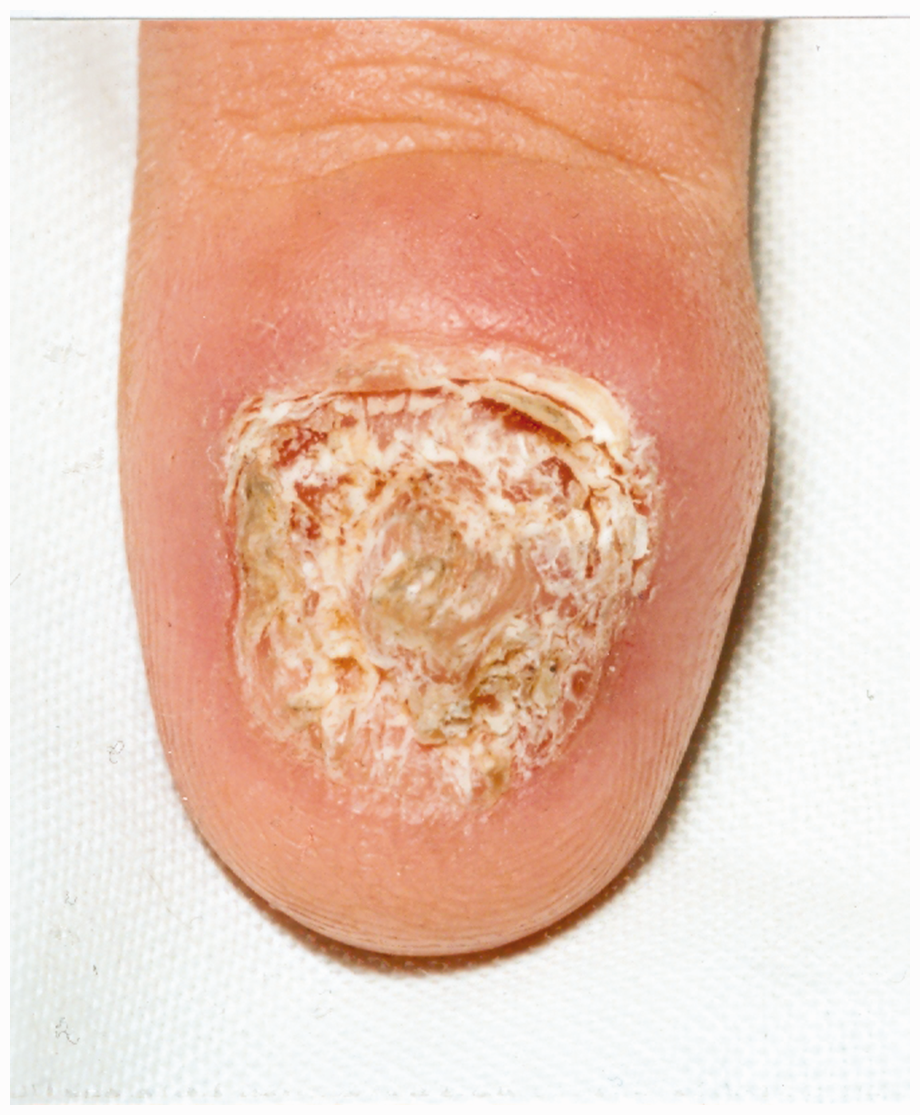
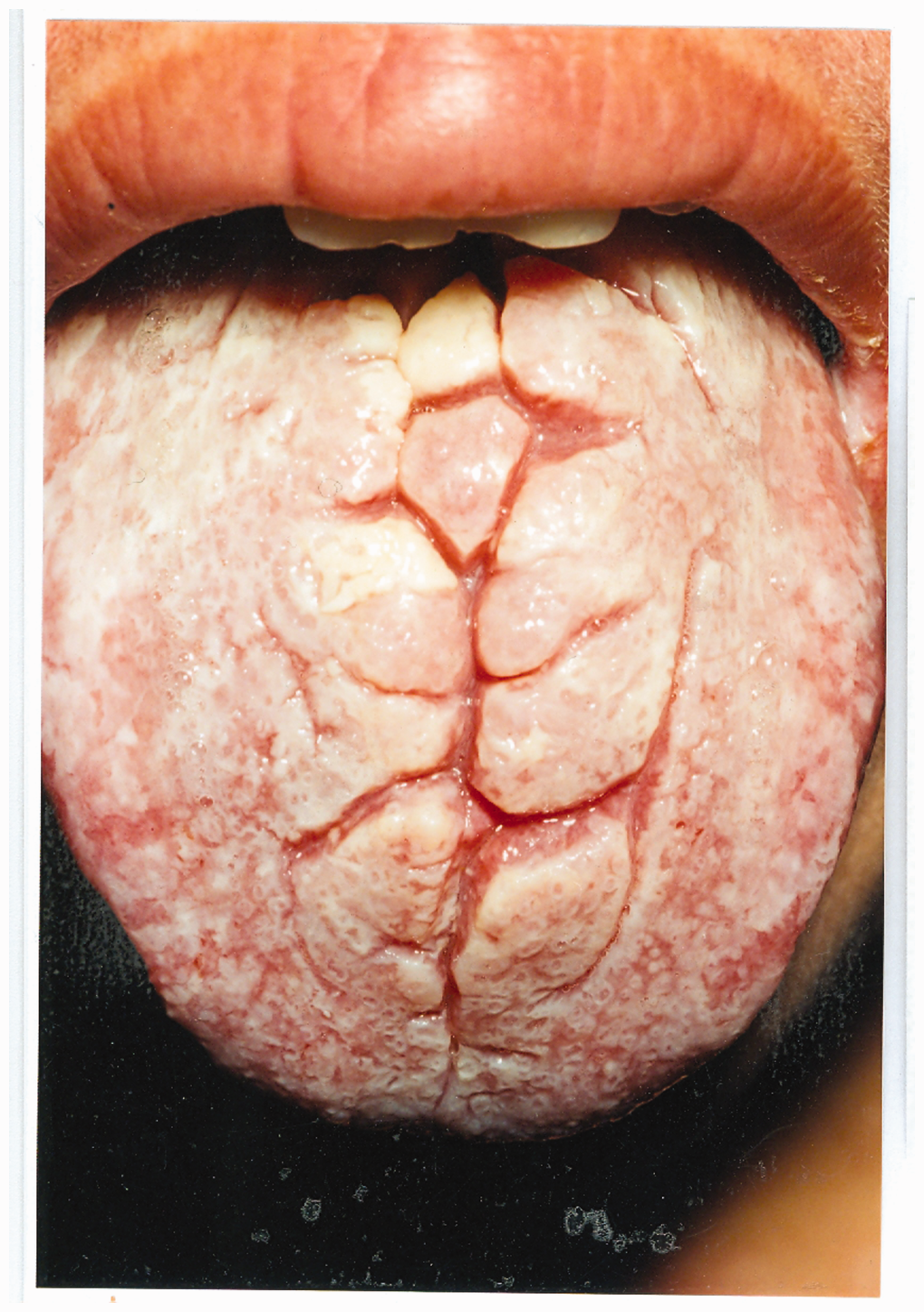
Case series
Our index patient (pt 1) was a mother of five children. She had suffered since childhood with severe CMCD presenting as oral, oesophageal and genital candidiasis and multiple, severe nail involvement as well as gastroesophageal reflux and difficulty swallowing, hypothyroidism, keratitis, blepharitis, conjunctivitis, styes, eczema, skin abscesses/boils and dental loss. At 58 years of age, she developed oral squamous cell cancer (SCC) which was treated with surgery and radiotherapy but lead to her death 3 years later.
Of her five children, three had CMCD. The eldest was a son (pt 2) who presented in childhood with severe CMCD, recurring lung disease with bronchiectasis, chronic obstructive pulmonary disease (COPD) and widespread eczema, skin infections, abscesses and boils. As an adult, he had intermittent oral thrush which was treated with anti-fungals (Figure 2). He also suffered with indigestion, dysphagia and odynophagia which responded well to proton pump inhibitors. At the age of 40 he had a gastroscopy for persistent symptoms which revealed marked oesophageal candidiasis as well as widespread oesophageal squamous cell carcinoma. Immunosuppressive chemotherapy was avoided in view of his underlying PID and instead he received palliative radiotherapy for extensive bony metastases, but passed away 6 months after diagnosis. The second sibling was his sister (pt 3) aged 34, who presented with chronic oral thrush since childhood which was treated with daily antifungals but nevertheless went on to developed severe dysphagia. Gastroscopy revealed stricturing of the oesophagus and the procedure was abandoned due to poor tolerance. She declined further investigations or surveillance. The third sibling (pt 4), who also had CMCD since childhood, was a younger brother aged 32, with intermittent oral thrush who underwent an initial gastroscopy at 25 years old, which revealed erosive oesophagitis. A repeat surveillance gastroscopy 7 years later was normal, with oesophageal biopsies showing mild oesophagitis with no dysplasia or candidiasis.
Clinical findings in CMC patients with GOF-STAT1 mutation A267V.
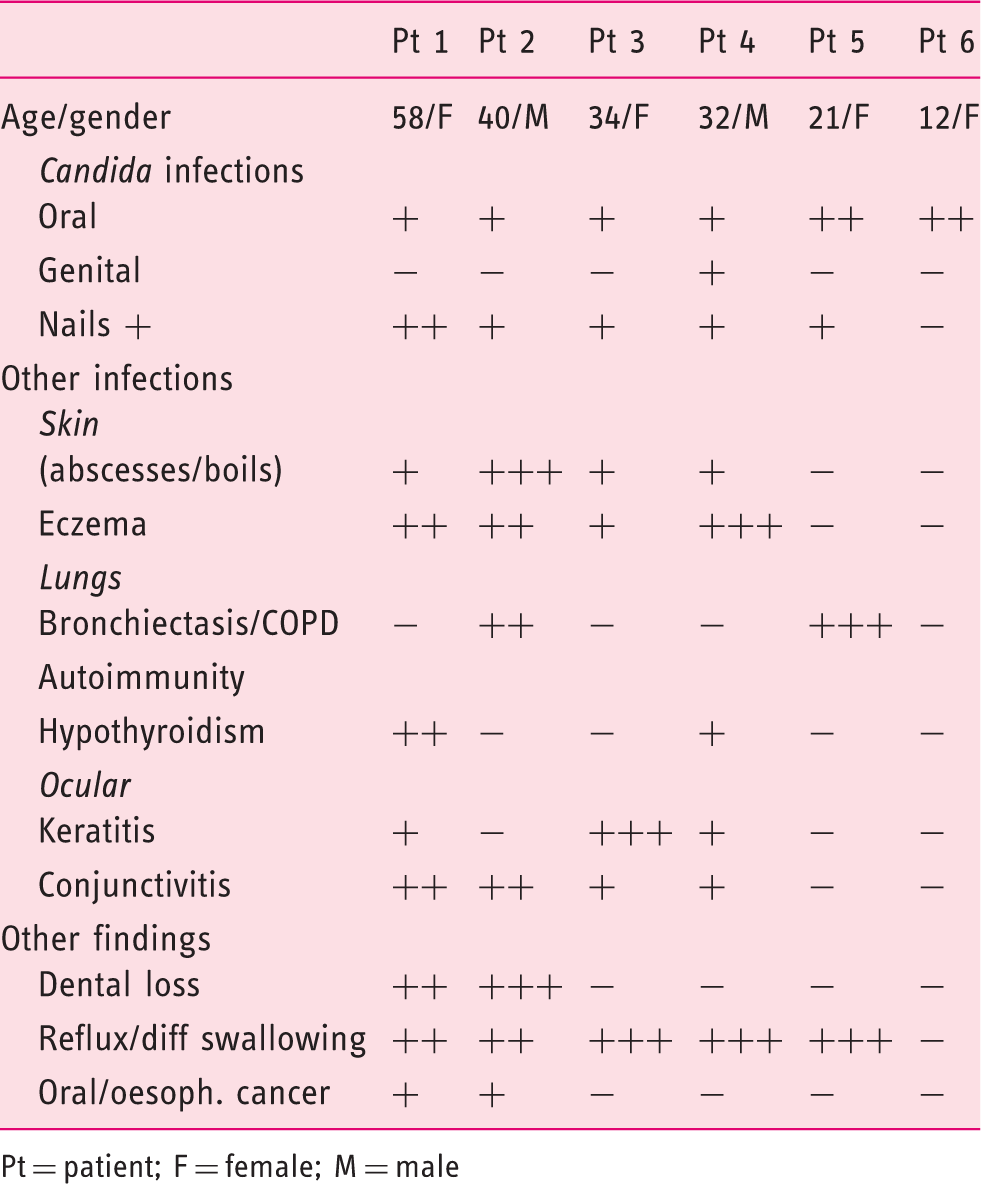
Pt = patient; F = female; M = male
Immunological findings in CMC patients with GOF-STAT1 mutation A267V.
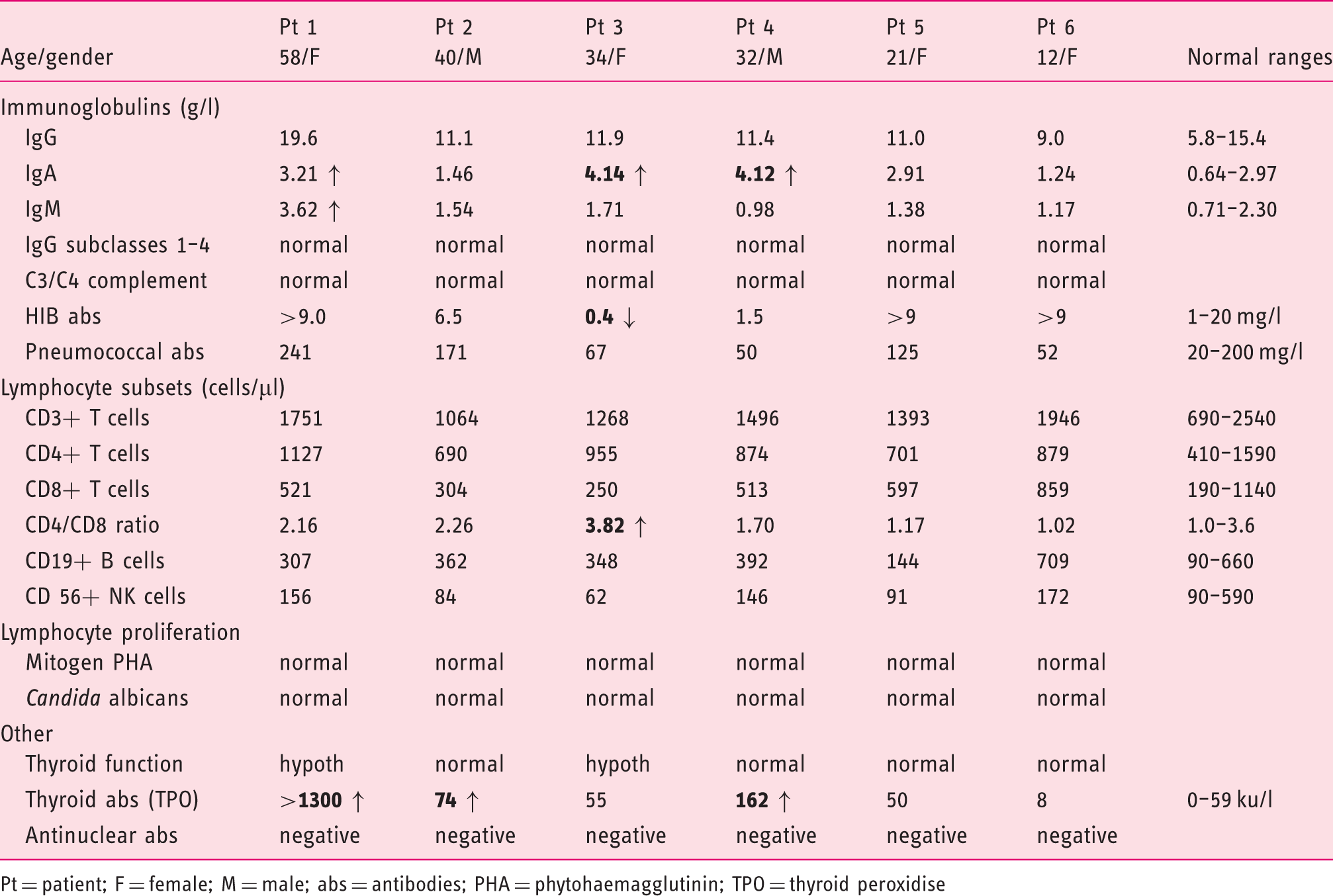
Pt = patient; F = female; M = male; abs = antibodies; PHA = phytohaemagglutinin; TPO = thyroid peroxidise
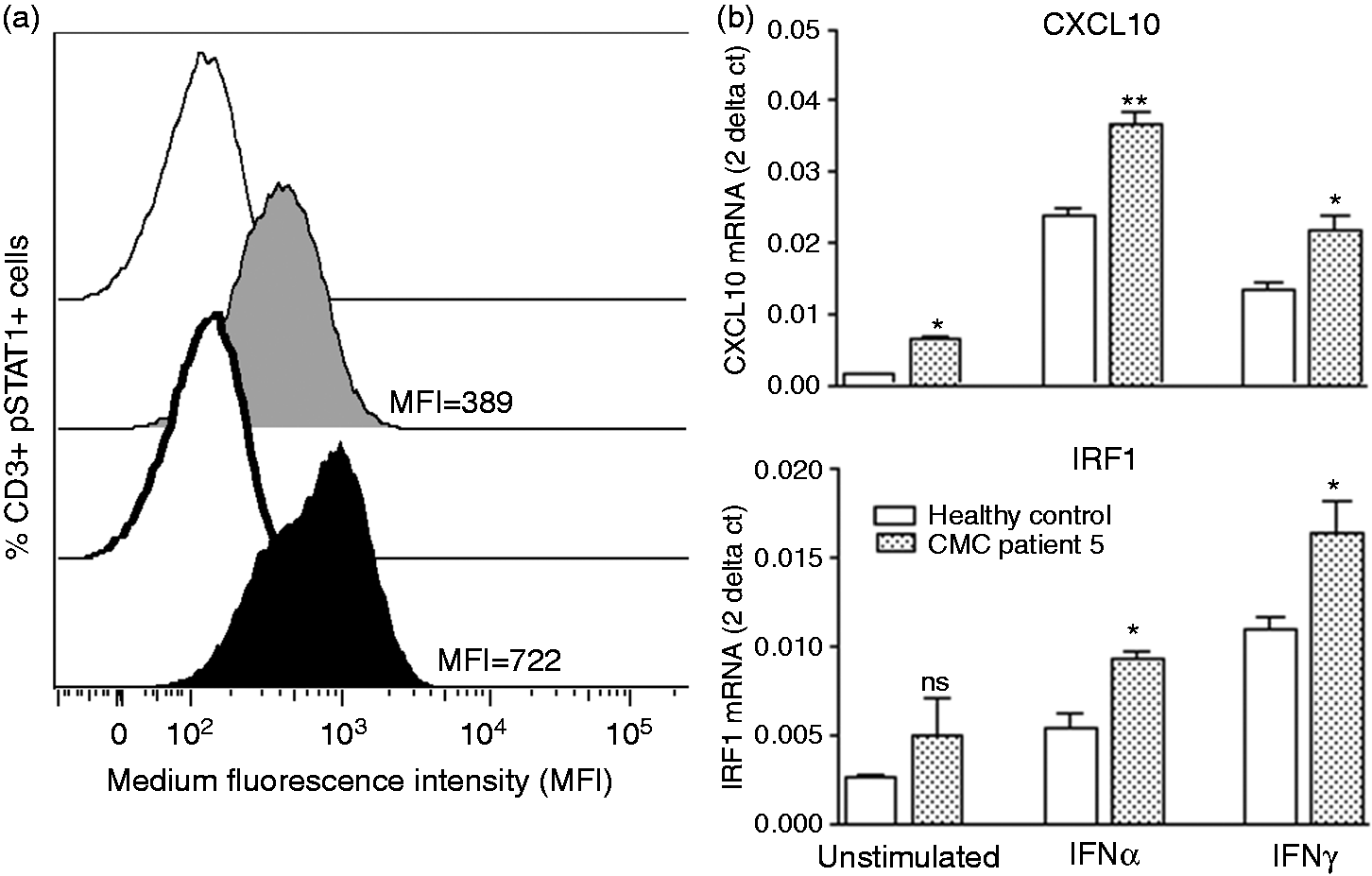
STAT1 gain-of-function in CMC patient 5. (a) STAT1 phosphorylation following stimulation with interferon (IFN) was assessed by flow cytometry. Whole blood from a healthy control and CMC patient 5 was stimulated with 103 IU/ml interferon (IFN)α (IFN-α2b Intron A, Merck Sharp & Dohme) for 30 minutes, cells were fixed, permeablised and stained with Alexa Fluor R 647 STAT1 (pY701) for pSTAT1 and CD3 Pacific Blue antibody for CD3 + T cells (all reagents from BD). Data were collected with a FACSCaliber (BD) and analysed with FlowJo software. Grey = healthy control; black = CMC patient; clear peaks = unstimulated cells; full peaks = IFNα stimulated cells. Median fluorescence intensity (MFI) is shown. (b) qRT-PCR of STAT1-dependent gene expression for CXCL10 and IRF1. In a healthy control and CMC patient 5, peripheral blood mononuclear cells were isolated and cultured in RPMI medium for 4 h with 1 × 103 IU/ml IFNα (IFN-α2b Intron A, Merck Sharp & Dohme), 1 µg/ml IFNγ (R&D Systems). Total RNA was purified using RNeasy Mini Kit (Qiagen) and reverse-transcribed into cDNA. Gene expression analysis for CXCL1o and IRF1 mRNA was performed with custom TaqMan gene expression assays (ABI). Gene expression was normalised to the housekeeping gene 18S rRNA. Data is expressed in the 2 − Ct format, where Ct = Ct (target) – Ct (18S). Results are for n = 3 replicates. Statistical significance was assessed by unpaired, two-tailed t test using Prism software. * = p > 0.05; ** = p > 0.01.
Discussion
It is now recognised that patients with CMCD, particularly those with the GOF-STAT1 mutation, have a broad clinical phenotype, significantly reduced life expectancy and an increased incidence of SCC. 11 Management of these patients always includes antifungal therapy to control mucocutaneous cutaneous candidiasis in spite of which SCC develops, indicating the need to consider urgent gastroscopies in the symptomatic patient as well as regular annual surveillance endoscopy with Lugol’s iodine chromoendoscopy to detect squamous dysplasia.
Unlike conventional oesophageal carcinoma, patients who develop oesophageal SCC on a background of CMCD present at a younger age. Delsing et al. reported a 25-year-old female who presented with dysphagia on a background of CMCD and GOF-STAT1 gene mutation, where the initial gastroscopy revealed only candidiasis, but the cytology confirmed malignant epithelial cells. A repeat gastroscopy 2 weeks later identified an irregular stenosis at 26 cm, and its histology was in keeping with SCC. Due to limited disease, the patient went on to have an oesophagectomy with reconstruction and colonic interposition with no further recurrence of oesophageal candidiasis or malignancy 20 years later. 10
The pathogenic mechanisms leading to SCC in the presence of CMCD are not fully understood, but it is believed to be due to chronic inflammation caused by the infection, as well to candidial catalytic activity leading to the production of nitrosamines such as nitroso-N-methylbenzylamine (NBMA) which is carcinogenic. 13
It is well known that the STAT1 gene is integral to innate immunity and mediates signalling of important inflammatory cytokines such as interferons (IFN) gamma (γ) and alpha (α). STAT1 is involved in mediating antibacterial, antiviral and antifungal immune responses, suppression of cell growth, and apoptosis as well as tumour genesis and tumour suppression. 14 Some reports have found its levels to be reduced in transformed cancer cells, suggesting tumour suppressor properties, which is also supported by Zhang et al. who found that transfection of STAT1c into oesophageal squamous cell carcinoma cells resulted in apoptosis and cell cycle arrest, while the absence of STAT 1 was associated with poorer outcomes. 15
Due to the high risk of oesophageal SCC in patients with CMCD, these patients should have an endoscopy if they have any upper gastrointestinal symptoms. They should also have regular surveillance endoscopy with chromoendoscopy using Lugol’s iodine or narrow band imaging to detect squamous dysplasia or early cancers. These patients present at a younger age compared to the conventional age for presentation of sporadic oesophageal SCC. Although the age at which this surveillance should start is not clear, it should be at least a decade earlier than the age of youngest member of the family diagnosed with oesophageal cancer. The intervals for surveillance endoscopy are also not certain, but at least annually or biannually. Squamous dysplasia of the oesophagus can now be treated endoscopically by endoscopic resection and radiofrequency ablation. 16
In view of the fact that the underlying mutation has been identified as a GOF-STAT1, new therapeutic approaches should also be considered such as inhibition of STAT1 signalling by specific inhibitors of the JAK-STAT signalling pathway. Obviously, the potential deleterious effects of inhibiting a crucial inflammatory pathway must be weighed against potential benefits, but it is encouraging to know that sporadic case reports have reported significant benefits without major side-effects. 17 However, this area still requires significant research.
Conclusion
In patients who present with recurrent oesophageal candidiasis we advise taking a thorough history, focusing on elements suggesting a PID such as age of onset, involvement of other body areas (skin and nail candidiasis), other infections, past medical history and family history of candidiasis. Additional testing should always exclude HIV infection as a cause of secondary immune deficiency. Consultation and/or referral to clinical immunology colleagues for further investigation and subsequent management of underlying PID is indicated.
CMC has a strong association with oral and oesophageal SCC, as such, these patients should be routinely monitored and investigated urgently if they present with upper gastrointestinal symptoms to detect early oesophageal cancer.
Footnotes
Conflict of interest
None declared.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.