
Abstract
Cystic fibrosis (CF) is one of the most frequently occurring inherited human diseases caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) which lead to ample defects in anion transport and epithelial fluid secretion. Existing models lack both access to early stages of CF development and a coeval focus on the gastrointestinal CF phenotypes, which become increasingly important due increased life span of the affected individuals. Here, we provide a comprehensive overview of gastrointestinal facets of CF and the opportunity to model these in various systems in an attempt to understand and treat CF. A particular focus is given on forward-leading organoid cultures, which may circumvent current limitations of existing models and thereby provide a platform for drug testing and understanding of disease pathophysiology in gastrointestinal organs.
Introduction
Cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene and affects the lungs, liver, pancreas, and intestine.
1
The CFTR channel regulates chloride and bicarbonate secretion in a cyclic adenosine monophosphate (cAMP)-dependent manner but has also been described as modulating other ion channels such as the epithelial sodium channel (ENaC).2–5 In various epithelia and exocrine glands, impaired CFTR-dependent ion conductance disturbs electrolyte and fluid homeostasis. Generally, several mechanisms synergistically cause luminal pH acidification and the production of hyperviscous mucus in secretory epithelial cells: (i) defective anion conductance with reduced chloride dependent fluid secretion; (ii) increased surface liquid depletion; and (iii) altered bicarbonate secretion (Figure 1).2–5 This leads to recurrent and chronic airway infection and pancreatic tissue destruction of varying degree while in the liver a rarely diagnosed phenotype termed cystic fibrosis-associated liver disease (CFLD) becomes evident. CFLD summarises herein a spectrum of liver disease reaching from mere fibrosis to full-blown biliary cirrhosis with portal hypertension and all arising consequences.
Regulation of ion concentration in pancreatic ductal fluid. (a) Along the ducts luminal chloride is reduced due to Cl− absorption while reciprocally bicarbonate concentration increases in the pancreatic fluid. (b) Main anion channels in pancreatic ductal epithelium involved in Cl−, HCO3−, and fluid secretion.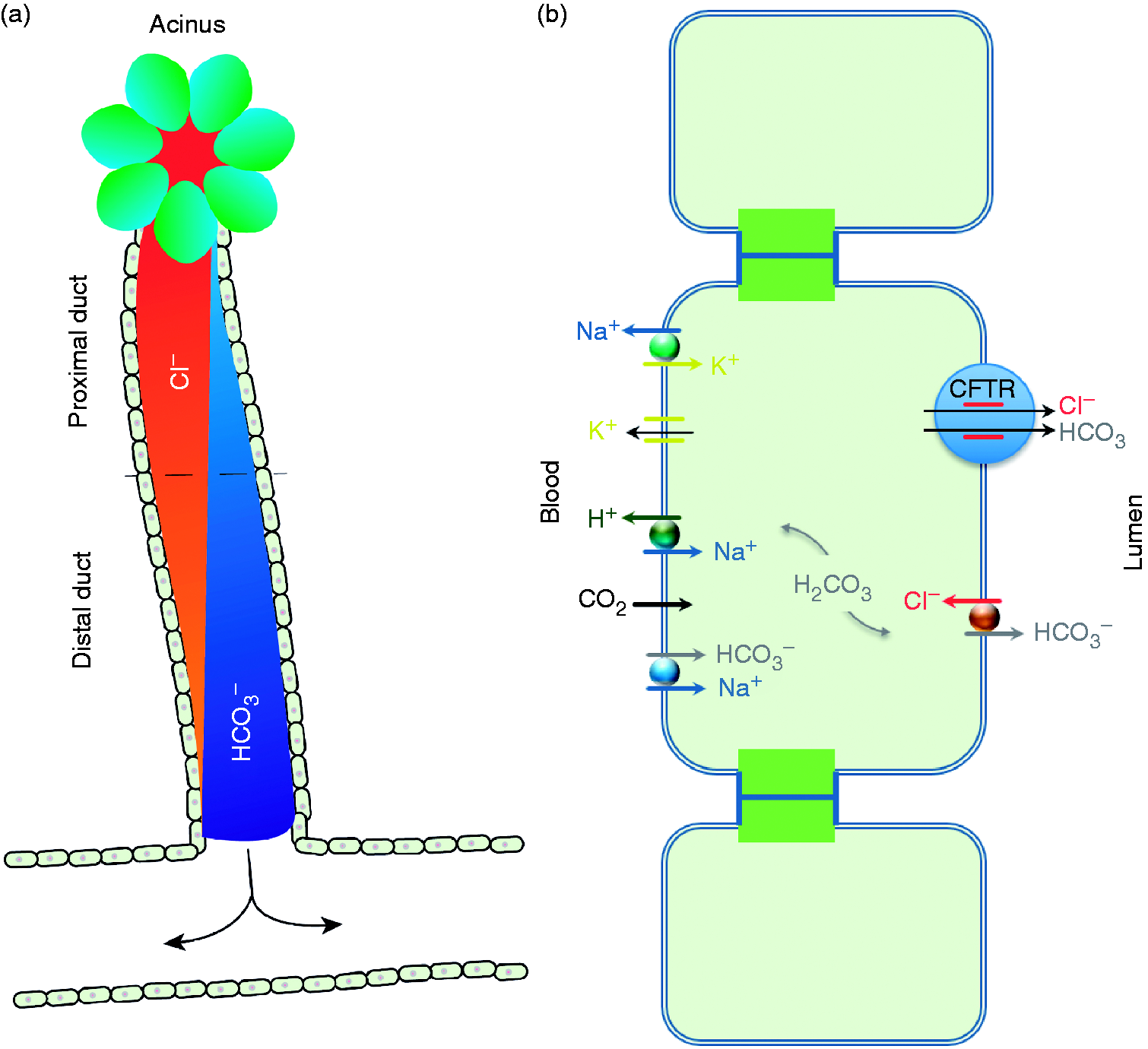
So far more than 2000 heterogeneous mutations (www.genet.sickkids.on.ca/app) have been described for the CFTR gene, located at 7q31.2., not all of which are clinically apparent. However, most of these gene variants lead to altered production, failed integration into the cell membrane, or limited function of the CFTR channel. CFTR mutations have been classified into six different types: while mutations in classes I–III establish a severe disease phenotype due to nearly complete loss of CFTR activity, class IV, V, and VI mutations have a more variable phenotype caused by residual activity.6,7
The most stringent correlation of the disease severity with the six-class system can be found in the pancreas. Here, certain mild CFTR genotypes have been shown to cause different likelihoods of developing pancreatitis, while class I–III mutations lead to exocrine insufficiency.3–5 The pancreatic fate of CF remains enigmatic as most research focuses on pulmonary CF, 8 and the need for a deeper understanding of the underlying molecular mechanisms takes a back seat due to the success of pancreatic enzyme supplementation and insulin therapy. 9
Indeed, chronic lung infections remain the primary cause of mortality, 1 despite successful interventions that dramatically improved survival not only due to medical, conservative management but also to improved success rates in lung transplantation. 10 Specifically, a recent study reported a continuous increase in the mean age of death from 1994 to 2010 in Europe, while also revealing a slightly poorer survival rate for females. 11 In 2014, it was the first time that the CF registry data base contained more adults than children with an increase from 29.2% in 1986 to 50.7% in 2014. This improved life span goes along with recent epidemiological data from a 20-year nationwide study in the United States that suggests a markedly increased cancer risk, particularly colon cancer. 12 In summary, gastrointestinal (GI) manifestations of CF become increasingly important due to improved patients’ life span. Thus, new model systems are warranted to mimic also non-lung complications of CF.
Most of the existing CF mouse models fail to fully reflect all important aspects of human CF, 13 and none allows the investigation of primary and living human patient material on a functional basis. Post-mortem analysis is restricted to morphological analysis due to fast autodigestive processes of the pancreas and developmental aspects also cannot be elucidated. The epigenetic context of a diseased individual is also not mirrored in any animal model. Organoid models from different sources may overcome most of these hurdles due to a series of advantages above current systems. The capability to provide a three-dimensional (3D) structure closely mimicking complex organs offers several advantages: (i) organoids are highly expandable, (ii) can form virtually every tissue, (iii) can recapitulate malignant transformation, and (iv) undergo successful transplantation and maturation in vivo as shown at least for some tissues such as the intestine. 14 In turn, this review discusses the current state of knowledge on the gastrointestinal facets of CF with a particular focus on various established organoid systems and their value to understand and treat CF.
The GI fate of CF
Clinical characteristics of CF within the GI tract
During the last years gastroenterologists are increasingly encountering patients with CF. Patients’ life expectation is continuously increasing and the disease, once considered as a burden of children and adolescents, is now showing several new facets many of those also affecting the GI tract. In general, the scientific knowledge about CF manifestations within the GI tract is limited because the predominant and mostly limiting infestation of all organs takes place within the respiratory tract. From the gastroenterological point of view primarily three different manifestations of CF can be highlighted: manifestations of CF within the (i) exocrine pancreas, (ii) hepatobiliary tract, and (iii) intestine. Here, pancreatic exocrine insufficiency plays a key role for the patients’ digestive dysfunction. Reduced secretion of bicarbonate and chloride leads to a more acidic and viscous luminal content, an observation which is not restricted to the pancreatic epithelium but can also be observed within the intestine and hepatobiliary epithelium. 5
Pancreatic manifestation
The most frequent cause of exocrine pancreatic insufficiency in children and adolescents is CF. The CFTR protein that functions as a cAMP-dependent chloride channel mainly regulates the passage of various anions at the apical membrane but also secretion of water into the pancreatic duct system. Thereby its activity enables the maintenance of an alkaline pH and sufficient secretory volume, which is essential to preserve the proteins secreted from the acini within a soluble state. Upon processing of pancreatic fluid, acinar cells initially secrete isotonic fluid mainly containing sodium chloride. Ductal secretion of bicarbonate within the pancreas is initiated in the proximal duct: Intracellular accumulation of bicarbonate leads to an osmotic secretion of the anion in exchange with luminal chloride (Cl−). In this process, CFTR exhibits recycling Cl− channel activity and plays an indispensable role for the activity of the apical Cl−/HCO3− exchanger. The net HCO3− transport drives efflux of water followed by increasing enrichment of the pancreatic fluid with HCO3− in the distal ducts due to a change in CFTR permeability for this anion. 15 Accordingly absence or reduced function of the CFTR protein results in diminished secretion of a more viscous and more acidic pancreatic exudate (Figure 1). This causes precipitation of proteins leading to a subsequent duct obstruction and pancreatic insufficiency. 16 However, the extent of pancreatic damage is dependent on the degree of CFTR malfunction. Therefore the severity of pancreatic disease varies between subclinical insufficiency (‘pancreas sufficiency’, PS) and severe pancreatic insufficiency (PI). CF patients with PI (90%) do not tend to develop chronic pancreatitis (CP) since a residual exocrine function is a prerequisite for auto-digestion. 17 The occurrence of severe pancreatic insufficiency is closely linked to the presence of two ‘severe’ alleles, on the other hand at least one mild mutation appears to be a prerequisite for PS in CF. 18 Thus, CP can only be observed in patients with a residual exocrine secretion as it is evident in class IV-VI CFTR mutations. The overall incidence of CP within CF collectives ranges from 1.5% up to 2%. 19 Interestingly, mutations within the CFTR gene can be found within a high proportion (42%) of patients with idiopathic pancreatitis. 20
Contrariwise, patients with PI (majority of CF patients) suffer from all consequences of exocrine pancreas insufficiency: steatorrhea and all facets of malabsorption, e.g. weight loss, oedema, anaemia due to vitamin B12 and folic acid deficiency, bleeding disorders (vitamin K malabsorption), metabolic bone disease, neurologic manifestations, and abdominal distension due to bacterial fermentation of unabsorbed food (Figure 2).
Impact of CFTR dysfunction in the gastrointestinal tract, hepatobiliary system, and the pancreas.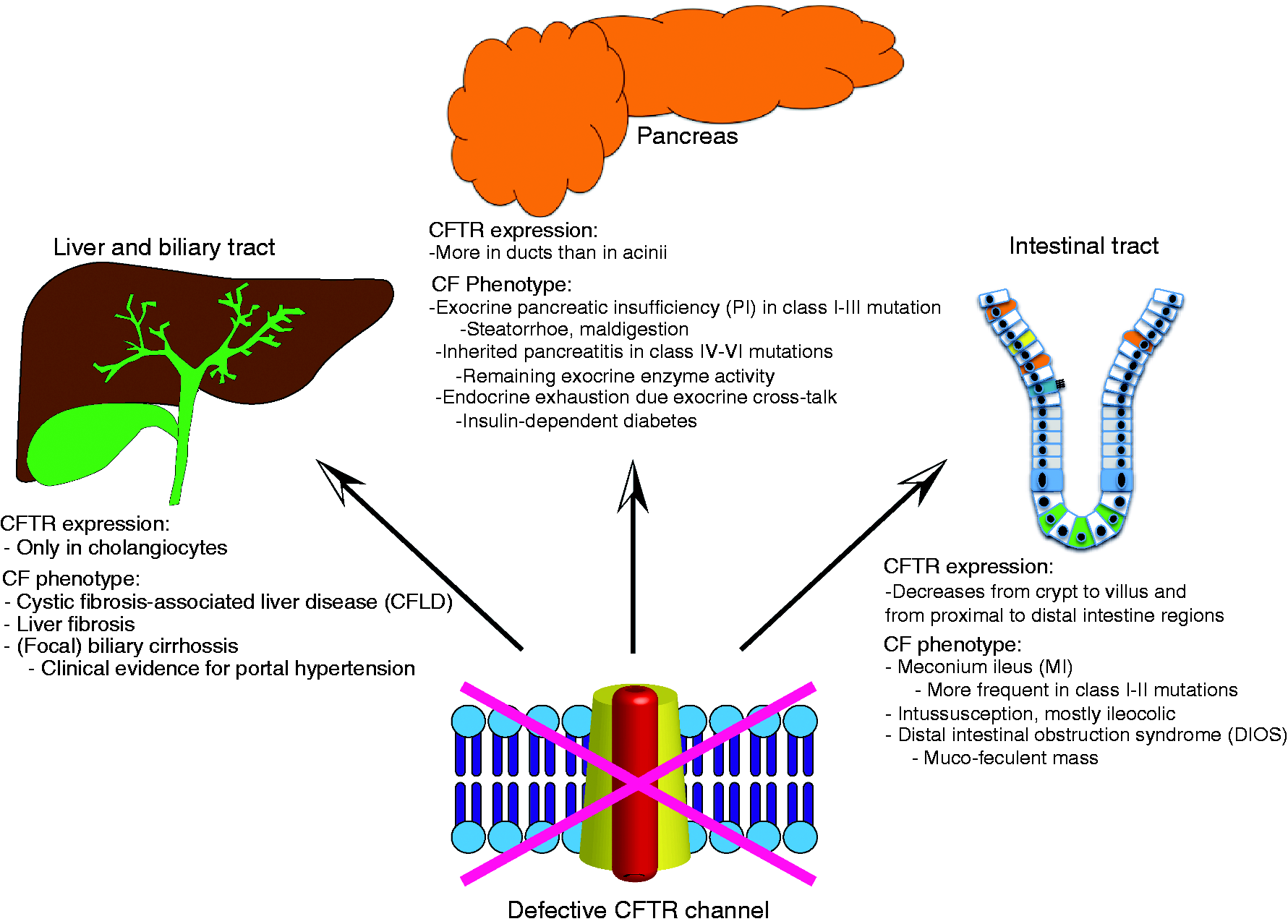
Approximately 30% of CF patients develop glucose intolerance. With the currently observed increase in life span more patients suffer from full-blown diabetes mellitus. In line, a low but continuously increasing fraction of patients develops diabetes-associated complications. Of note, the process of endocrine failure in CF is poorly understood but recent evidence suggests a negative influence of the damaged exocrine compartment on the islets of Langerhans. 21
Hepatobiliary manifestation
Within the liver CFTR is only expressed in cholangiocytes but not within hepatocytes. 22 Focal biliary cirrhosis is a key feature of hepatic manifestations of CF. A lack of CFTR function causes a more acidic and more viscous bile composition leading to an obstruction of bile ducts. 22 CFLD is the third most frequent cause of death in patients with CF. Analogously to pancreatic manifestation there is a wide range of liver affection in CF: clinicians observe both subclinical CFLD and severe forms with eventual organ dysfunction. Severe forms of CFLD are only present in about 5% of all patients with CF, in most of the cases they are linked to the presence of the SERPINA1 Z alleles. 23 Consequently, all kinds of complications of liver diseases, e.g. portal hypertension, ascites, reduced liver synthesis, hypoalbuminemia, coagulation disorders, infectious disease, and liver failure can occur within the course of CF (Figure 2).
Intestinal manifestation
Intestinal manifestations of CF result on the one hand from pancreatic insufficiency as described above but also from abnormal enteric fluid secretion: An epithelium-autonomous defect contributes to intestinal manifestations of CF by disturbed exocytotic dynamics in various intestinal cell types, e.g. goblet cells. 24 Upon impaired CFTR function, not only a reduced total volume of fluid secretion, but also an altered interaction between epithelial cells and the gut microbiome has been described. 25 A relevant portion of patients (up to 20%) shows the first clinical signs of CF within the first days of their life: meconium ileus. The formation of meconium ileus (MI) in neonates can be understood as a clinical evidence of pancreatic insufficiency and thus hints at a ‘severe’ CFTR mutation, as described above. 26 The prevalence of MI differs between different mutation types with class I and II harbouring an increased risk of MI formation compared to other class mutations. Of note, CFTR modifying genes have been described to contribute to MI.
Apart from this first paediatric intestinal manifestation of CF, knowledge about other acute intestinal complications of CF is relevant for gastroenterologists, such as:
Patients with CF are at a significantly higher risk to develop intussusception, this potentially threatening event takes place in up to 2% of CF patients.
22
Most of the patients suffer from an ileocolic intussusception, often confronting physicians with a challenging clinical presentation. In many cases high frequency sonography can verify the diagnosis. Spontaneous resolution of intestine invagination occurs in a fraction of patients. Another relevant intestinal complication of CF is the ‘distal intestinal obstruction syndrome’ (DIOS). About 18% of all patients with CF suffer at least once from a DIOS: an adherent ‘muco-feculent’ mass leads to an intestine obstruction posing the risk of a mechanical ileus. A differentiation between ‘conventional’ obstipation and DIOS might be difficult. DIOS can be diagnosed with a characteristic appearance in abdominal x-ray. A differential diagnosis between ‘appendicitis’, DIOS, and intussusception might also be a challenge, especially as CF patients often show an increased diameter of the appendix while having an overall lower incidence of appendicitis compared to non-CF controls (Figure 2).
27
CFTR mutations and cancer
The overall lifetime risk for the development of most cancers does not differ in CF patients compared to the standard population. However, it has become evident that – alongside with an increased life expectancy due to better treatment regimen and possible organ transplantation – the incidence of certain malignancy increases in patients suffering from CF. This is most evident for cancers arising from the digestive tract: Malignancies of the oesophago-gastric junction, the biliary tract, but also small bowel and colon are significantly more frequent in CF patients compared to the general population. 12 This might be due to the relatively higher CFTR expression in the gastrointestinal epithelium. CF has also been identified as a contributing hereditary component in pancreatic ductal adenocarcinoma (PDAC). Homozygous CFTR gene mutations bear a 5.3-fold increased risk of PDAC, of note in relevantly younger patients (35 years median).28,29 In contrast, heterozygous CFTR gene mutations are closely connected to chronic pancreatitis,30,31 an independent risk factor for PDAC development. 32 Mutations in the CFTR gene are not exclusively associated with PDAC, moreover they are found in several cancers, mainly of gastrointestinal origin, like colon or gastric cancer,12,20,33–35 but also extra-gastrointestinal like ovarian, prostate, and non-small cell lung cancer.36–39 The specific role of CFTR mutations in various malignancies has still to be elucidated, particularly in light of the fact that the CFTR gene locus is commonly hypermethylated.38,40
As far as CFTR is linked to epithelial cell polarity and integrity of tight junctions, a requirement for the intact barrier function, alterations can be linked to processes evident in malignancies like epithelial–mesenchymal transition (EMT). 41 In human breast cancer cells the inhibition of CFTR induces TGFβ1 and promotes migratory and invasive properties. Here also the inhibition of the NF-κB signalling via urokinase-type plasminogen activator (uPA) seems to be relevant.36,42 In human breast cancer patients CFTR expression was significantly downregulated within the tumour samples and this was linked to a poor prognosis. 42 In colorectal cancer (CRC) cells, knockdown of CFTR reduces epithelial tightness, mainly linked to the interaction with AF-6/afadin. Similarly, in human CRC samples expression of CFTR and AF-6/afadin was significantly downregulated and correlated with a dismal prognosis. 33 Altogether the disrupted cell–cell adhesions through CFTR alterations seem to be important and the link to EMT a promising approach to follow.
In a recently published mouse model, CFTR was knocked-out in the intestinal epithelium resulting in a significantly increased number of intestinal cancers and colon adenomas, indicative of a tumour suppressor function. Interestingly, gene clusters found to be dysregulated in the CFTR knock-out mice belong to the immune response and the intestinal stem cell compartment in line with the observed inadequate immune response as a hallmark feature of pulmonary CF. However, inflammation is not restricted to the respiratory system but rather has systemic impact, 43 and particularly affects the pancreas and intestine.44,45 As far as chronic inflammation can maintain cancer development, this may be a further contributing factor.46,47 Knockdown of the CFTR gene also leads to a pro-inflammatory state, with up-regulation of inflammatory cytokines like IL-6/8, causing oxidative stress in intestinal epithelial cell lines. 48 The zinc-finger nuclease based abrogation of the CFTR gene in the same cells (CaCo-2/15 enterocytes) resulted in an increased inflammatory cytokine secretion and higher levels of the proinflammatory transcription factor NF-κB levels. Moreover increased lipid peroxidation levels and a reduced antioxidant defence confirmed by reduced enzymatic activities of glutathione peroxidase and catalase were observed. 49
In summary, there is currently no published data that reliably connect these chronic proinflammatory features with an increased cancer risk in CF patients, but the hypothesis seems to be of interest as this association has been demonstrated for various other chronic inflammatory diseases. New model systems such as the later on highlighted organoids may aid to understand this hypothesis.
Current GI models of CF
Applicability of various in vitro culture systems and animal models to investigate different aspects of cystic fibrosis.
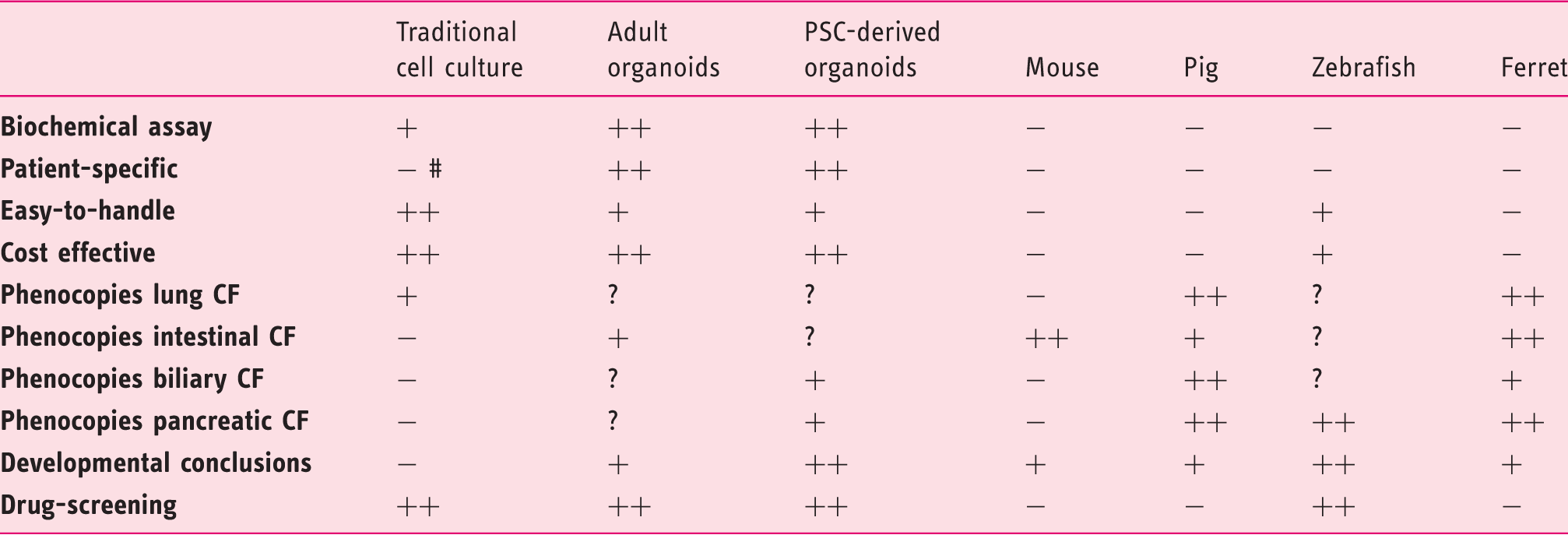
++ very high convenience; + high convenience; - low convenience; ? not investigated so far; # primary airway epithelial cell have been described.
Disease manifestations in small and large CF animal models.
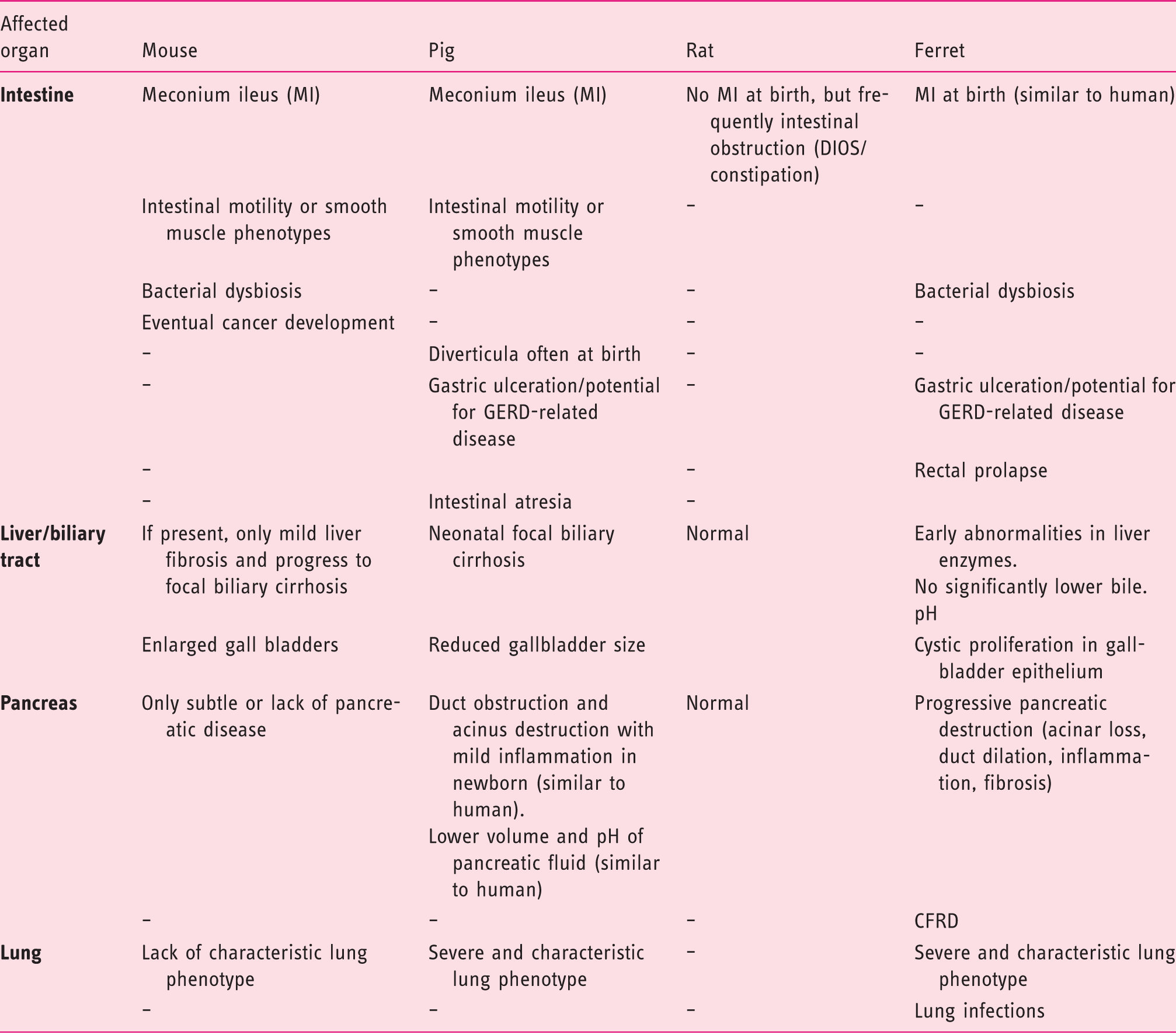
DIOS: distal intestinal obstruction syndrome; GERD: acidic gastroesophageal reflux disease; CFRD: cystic fibrosis related diabetes.
Traditional cell culture models
Studies in immortalised cell lines complemented by primary epithelial cells and isolated rodent ducts have contributed a lot to our knowledge about the role of CFTR in basic biochemical and physiological processes, specifically the consequences of its dysfunction for ion transport and secretion.15,55–59 Analysis of HEK293 cells transiently transfected with CFTR mutants specific for PS or PI patients found abnormalities in bicarbonate regulation as a cause for different pancreatic phenotypes. 15 These systems are still suitable to discover novel aspects of cellular impairment by mutant CFTR. For example, overexpression of F508del-CFTR in HeLa cells showed that its interaction with cytokeratin 8 may lead to the F508del protein processing defect and disruption of this binding could restore channel function. 60 Regarding intercellular junctions, a recent study suggested a negative effect on gap junction function based on mis-trafficking of connexins observed in F508del-CFTR expressing human airway cells. 61 Moreover, most high-throughput screens to identify small-molecule compounds that improve cellular processing (so-called ‘correctors’) and/or gating function (‘potentiators’) of F508del-CFTR have been carried out on recombinant cells.62,63 However, those studies primarily use human bronchial epithelial cells to evaluate selected candidate drugs, reflecting their design to rescue the lung-phenotype of CF while effects on pancreatic and gastrointestinal phenotypes of CF remain elusive.62–65 In addition, evidence is accumulating that the efficacy of correctors differs between cell lines and might be even lower in native tissues accounting for variable success in patients. 66 Thus investigations in more relevant cell types are required but have been hampered by limited availability of primary human tissue requiring endoscopic fine-needle biopsies (for pancreas) and other challenging methods for isolating and in vitro culturing of, for example, cholangiocytes or pancreatic cells.67,68 Some functional analyses of CFTR activity have been performed in human rectal mucosa biopsies and revealed highly variable (low to no) residual chloride secretory activity in the tissue of individual CF donors.69–71 Residual CFTR function was not only dependent on specific CFTR variants and correlated with a mild disease phenotype, 69 but even differed within patients all carrying the same homozygous F508del mutation. 70 These observations highlight the need for individualised drug screening approaches that do not just focus on few mutations and also take patient-specific factors into account such as epigenetics or co-founding mutations (Table 1).
Stem cell models
In recent years techniques for isolation and expansion of human intestinal as well as liver stem cells have advanced the field and will help to overcome limitations in availability of sufficient patient material.72–75 The development of the reprogramming technology represents another breakthrough, 76 now enabling researchers to generate induced pluripotent stem cell (iPSC) cultures from easily accessible patient cells such as skin fibroblasts or hair keratinocytes, 77 which can be differentiated into various specialised cell types, e.g. intestinal tissue.78–80 Patient-specific pluripotent stem cell models have been established for CF.81–83 By mimicking steps of lung development, CF-iPSCs could be directed to form mature airway epithelium in vitro that upon treatment with a CFTR corrector compound (C18, analogue of VX-809) exhibited enhanced plasma membrane localisation of mature F508del CFTR protein. 83 This demonstrates the potential of iPSC-derived epithelial cells as CF-disease models for therapeutic identification or validation of candidate drug effects, which we will discuss in more detail in a subsequent section.
Furthermore, site-specific genome editing tools, including TALENs, zinc-finger nucleases, or CRISPR/Cas9, offer the possibility to correct the target gene mutation for establishing isogenic control lines, create rare CFTR mutations or introduce a deletion in human adult as well as pluripotent stem cells. 84 Comparative analyses in relevant tissues derived from stem cells engineered in that sense would be less biased by the genetic background thus allowing to selectively dissect molecular and cellular causes for defects in individual CFTR variants on the one hand and the impact of potential genetic modifiers (genes otherwise accounting for heterogeneity in patient outcome 85 ) on the other hand. The first substantial approach in the light of CF was the CRISPR/Cas9-based correction of the CFTR locus by homologous recombination in cultured intestinal stem cells of a patient. In cultured organoids the corrected allele was proofed to be expressed and functional. 86 As proof of principle a recent study applied zinc-finger nucleases for mutant CFTR gene correction via homology-directed repair in patient-specific iPSCs, which restored channel function in iPSC-derived epithelial lung progenitor cells (Table 1). 87
Animal models
Animal models have been invaluable for studying CF pathogenesis and functional consequences of CFTR mutations on the whole organ system. However, the diverse CF animal models exhibit large differences in disease manifestation and progression and show variable penetrance of characteristic pathologies recapitulating human disease only incompletely. Nevertheless these models, especially the newer pig and ferret models, provide insights into early stages of disease development and help to understand the complex pathophysiology in the gastrointestinal system therefore allowing an advanced look on pancreatic and biliary CF fates.
While CFTR-deficient mice, 51 and lately created knockout rats, 52 lack the characteristic lung phenotype and obvious pancreas or liver defects, CF pigs and ferrets develop severe lung disease and are highly susceptible to airway infections similar to CF patients (Tables 1 and 2).53,54,89–91
Intestinal obstruction is even more pronounced in all four species compared to humans and can be lethal in many cases thus requiring prophylactic polyethylene glycol administration to CF pigs and ferrets.89,92 However, to circumvent meconium ileus in newborn F508del or CFTR-/- piglets surgery is frequently necessary, which allows to practice surgical techniques for this kind of clinical problem.88,89 The severe intestinal phenotype has been alleviated by transgenic CFTR expression from the intestinal fatty acid-binding protein (iFABP) promoter to restore channel function specifically in the intestine without pancreatic or hepatic correction in pig and ferret lines, hence these animals still suffer from lung, pancreas and liver disease.90,93
CF pigs show hepatic lesions similar to those in humans with CF, most strikingly neonatal focal biliary cirrhosis but also microgallbladder, 94 and have abnormal, more acidic biliary secretion. 95 Although newborn CF ferrets display abnormalities in liver enzymes, older animals have variable hepatic pathologies dominated by manifestations in the gallbladder such as cystic mucosal hyperplasia.90,92 In the future these models would be helpful for developing preventive treatment and diagnostic strategies to manage liver disease in CF patients at an early stage. 96
Pancreatic damage in pigs with CF closely resembles the human disease with beginning in utero, acinar cell destruction and inflammation in newborns and lower volume and pH of pancreatic fluid.95,97 Comparison of 83- to 90-day-old foetal (birth after about 114 days of gestation) and newborn pigs revealed a progressive loss of zymogen-filled mature acini and inflammatory cell infiltration. Moreover gene sets for inflammatory response such as response to injury and host defence response, as well as for complement cascade and induction of apoptosis were enriched in pancreata of foetal and newborn CF pigs suggesting that proinflammatory, proapoptotic, and profibrotic pathways are actively involved in disease initiation while tissue remodelling like fibrosis or metaplasia seemed to be a response to the pathogenic process. 97 CFTR deficient ferrets exhibit similar features although less severe at birth. 90 Therefore abnormalities in blood glucose and insulin regulation in the ferret model can be examined before the destructive process in exocrine tissue accompanied by endocrine pancreatic loss advances, 98 which is useful for understanding CFRD (CF-related diabetes) in CF patients. 99 Apart from the mammalian CF models, the recently described CFTR mutant zebrafish, 50 which displays many pancreatic changes similar to CF in humans, is a very promising model for studying the onset and sequence of events leading to rapid acinar tissue destruction. Investigations of early larval stages led to the conclusion that loss of CFTR function does not impact early pancreas development but rather impairs pancreatic duct function initiating organ destruction after its formation. 50 Another beneficial feature of the zebrafish model is the possibility to perform straightforward screens for genetic and chemical CFTR modifiers in vivo. 100
Overall particularly the pig model seems to be most suitable to further analyse pathophysiological mechanisms of CF and to evaluate potential therapeutics in vivo since it reflects many gastrointestinal and pancreatic aspects as well as the lung phenotype of human CF, which cannot be mirrored in CFTR deficient mice. Nevertheless, we can just as well benefit from the species-specific discrepancies regarding disease manifestation: Thus, a recent report provides an explanation for impaired respiratory host defence in CF pigs and humans and the obvious protection from chronic airway infection in CF mice. The authors narrow it down to imbalanced H+ secretion by ATP12A (non-gastric H+/K+ ATPase) causing defective bacterial killing due to airway acidification in pigs and humans in contrast to mice that express much less ATP12A. 101 This demonstrates how cross-species comparative studies could help to understand mechanisms of CF pathogenesis and to identify new therapeutic targets (Table 2).
Organoid culture as a multipurpose platform to model CF, GI development, and cancer
The invention of culture conditions for long-term expansion of even a single LGR5+ intestinal stem cell as crypt-villus structures demonstrated the feasibility to derive complex organ-like structures in vitro from primary adult tissues and initiated a wave of studies creating epithelial organoids from other endoderm-derived organs.72,74,75,102–105 Human intestinal organoids or ‘mini-guts’ are grown in a 3D culturing system in matrigel and supplied with a specific set of growth factors such as R-SPONDIN 1, WNT3A, EGF, and NOGGIN.74,106 In this environment, isolated adult stem cells build crypt-like structures composed of diverse organ-specific cell types recapitulating human tissue architecture and the associated stem cell hierarchy responsible for rapid epithelial turnover and tissue homeostasis in the gut.74,107 Therefore these organoids are not only suitable for disease-modelling and drug validation, but also for studying tissue development, regeneration and niche organisation of the GI tract (Table 1). Notably, researchers succeeded in deriving intestinal organoid cultures from human pluripotent stem cells (hPSCs) by mimicking intestinal lineage commitment during embryonic development. Growth factor-stimulated guided differentiation of PSCs through the definitive endoderm, primitive gut tube and mid-/hindgut stage results in the formation of spheroid structures that undergo morphogenesis to form a functional gut.78,79,108,109
For the purpose of modelling CF in vitro, Dekkers et al. established an assay implementing patient-derived intestinal organoids. 110 Making use of the cyst-like organoid structure, which is constituted by a columnar epithelium surrounding the lumen, the authors show that forskolin-induced CFTR-mediated ion transport at the apical membrane of the epithelium results in swelling of the organoid (from isolated mouse and human crypts) due to increased fluid secretion into the lumen. In organoids established from CF patients this effect is strongly reduced but can be restored by treatment with CFTR corrector and/or potentiator compounds.110,111 Hence this system can be used to analyse potential drugs for their capability to rescue the disease phenotype in a patient- and tissue-specific manner. Only recently proof of concept has been provided by showing that rectal biopsy-derived organoid swelling in vitro correlates with individual response to drug therapy in vivo allowing for preclinical selection of responders to CFTR modulators. 112
The attempt to transfer this idea to other affected GI organ systems has been hampered by the lack of primary human pancreatic or biliary tissue and appropriate culture techniques. Recently the isolation of stem cell populations giving rise to self-organising organoids from biopsy samples, e.g. after fine needle aspiration during endoscopic ultrasound, 74 has also been reported for human liver, pancreas, and premalignant lesions in the GI tract.75,105 These adult stem cell derived epithelial organoids are of particular value since their long-term stable expansion and multipotentiality offer the possibility to investigate the tissue-specific stem cell characteristics and differentiation potential. Moreover analysis of molecular pathways in this context may contribute to our knowledge on how to manipulate cell fate specification or to induce de- or transdifferentiation.
Nevertheless, human 3D organoid cultures are challenging and mostly require supplementation with the expensive LGR5-ligand R-SPONDIN 1 to maintain self-renewal of the stem cell population. More importantly, the procedures to obtain tissue material like puncture of the pancreas or liver biopsy are difficult to perform, cumbersome, and always harbour a risk for the patients, which restricts their routine applicability.75,105
Instead, hPSC-based models could be generated to assess pancreatic and biliary phenotypes of CF. Therefore our group and others have designed an approach to direct hPSCs towards pancreatic progenitor cells that form organoids composed of functional acinar- and duct-like structures under 3D culture conditions. Applying this tool to iPSCs derived from CF patients we show that pancreatic cell specification, achieved by recapitulating developmental events in vitro, occurs basically normal. 113 This observation provides evidence that loss of CFTR does not impair early pancreas development in humans and is supported by results obtained in the zebrafish CF model. 50 Importantly, CFTR mutated pancreatic organoids faithfully mirror the patients’ phenotype in the functional swelling assay and thus display a novel disease model for individualised drug testing specifically in pancreatic tissue. 113
To model biliary diseases, Sampaziotis et al. and Ogawa et al. in parallel developed a protocol for the stepwise differentiation of iPSCs into cholangiocyte-like cells (CLC).114,115 The cystic organoids that formed in 3D culture displayed functional characteristics of biliary epithelial cells such as enzymatic activity (ALP, GGT) and response to secretory stimuli. CFTR activity in healthy-donor derived CLC organoids was confirmed by monitoring changes in intracellular and intraluminal chloride concentration with a fluorescent chloride indicator in response to different chloride challenges. Analysis of CF patient-derived organoids carrying the F508del CFTR mutation revealed the loss of CFTR functionality in this assay while treatment with the CFTR corrector VX809 (lumacaftor) could restore CFTR-mediated intraluminal chloride secretion.114 In the second study the correctors VX809 and Corr-4a in combination with the potentiator VX770 were successfully applied to improve defective swelling of cholangiocyte cysts from CF-patients.115 To conclude, the authors provide a promising iPSC-based system for modelling CF-associated cholangiopathy and for investigating the effect of novel drugs in a tissue that we otherwise lack access to. 114 Only recently ivacaftor (VX770) was approved by the US Food and Drug Administration (FDA) and the European Commission for the treatment of CF in an eligible group of patients carrying the G551D-CFTR mutation, 64 nine other class III variants, 116 or a class IV mutation (R117H-CFTR).117,118 A combination therapy of VX770 and VX809 had only moderate but at least significant beneficial effects in clinical trials,119,120 and has been approved as ‘the first medicine to treat the underlying cause of CF’ in patients homozygous for the F508del-CFTR mutation.117,121 To our knowledge, all current drugs have been primarily designed to rescue the lung-phenotype of CF,116,119,120,122 while the effect on pancreatic or bile duct disorders in CF patients have not yet been explored. Although it is currently unclear to what extent results obtained in the organoid systems are transferable to individual patients, another key advantage of the organoid system is the capacity to generate virtually every organ derived from a pluripotent stem cell. This opportunity is particularly important as CF phenotypes may have organ-specific differences but also patient-specific differences. Thereby, the evaluation and comparison of the CF phenotype becomes possible across various organs despite being in the same genetic background. This will allow us to develop and test organ-specific drugs being tailored to cure the patients’ maximum phenotype in the GI tract but also the lung. Also, large animals are cost intensive and long generation times prevent large-scale production of the diseased animals.
But also in the field of cancer research, organoid culture is increasingly applied. In the light of disease-associated risk for the development of GI malignancies in CF, patient specific tumour-organoids might incorporate a tool for advanced disease modelling of CFTR associated tumorigenesis. The most frequently used models to recapitulate and study human solid cancers are cultured cancer cell lines and patient-derived xenografts that have certain limitations. Cancer organoids display a new powerful tool in cancer research.105,123 Regarding PDAC there are already established cancer organoid models derived from human cancers or recently from hPSCs.105,124 Considering special 3D culture conditions, hPSCs form exocrine progenitor organoids in which ectopic expression of mutant KRAS or TP53 is sufficient to induce mutation-specific phenotypes in vitro and in vivo. 124 Moreover, it could be shown that the genetic diversity (means the genomic and epigenetic state) is preserved in cancer organoids cultured from biopsies of human CRC metastases. 125 Besides, the fact that the quoted organoid systems can recapitulate the basic cancer biology and the influence of selected mutations makes them promising for drug screens (see below). Gene editing systems, particularly CRISPR/Cas9, are a powerful tool to improve the specificity of (cancer) organoids.
Organ-specific drug screenings in organoid systems – hype or hope?
As outlined above, the last 5 years initiated a wave of newly established organoid systems from man and mice that has brought hope to novel human therapeutic approaches. Up to now we have to ask the question how we will be able to apply these novel techniques to translate them into therapeutic strategies. We hypothesise that especially in the gastrointestinal and pulmonary disease fields organoid model systems will be and partially are already the nearest future for drug screenings as well as personalised medicine. But other organ systems will follow soon. Although, so far, no industrially applied large-scale screenings in organoid systems are published, the road is already paved. In 2015 an outstanding study addressing ductal pancreatic cancer modelling showed that exocrine pancreatic organoids can be established from hPSCs by guided differentiation. Furthermore by ectopic expression of mutant KRAS or TP53 in these progenitor organoids PDACs can be realistically modelled in vitro. As a proof of principle and direct comparison to the established cancer model, organoids from fresh biopsies of PDAC patients were generated and totally resembled the patients’ phenotype. Morphology and molecular comparison of the stem cell- and biopsy-derived organoids confirmed the validity of the established models. 124 Applied inhibitors for epigenetic regulators on the tumour organoids in combination with and without gemcitabine, as a standard treatment against PDAC, displayed a dose-dependent influence on proliferation and thereby further confirmed the similar epigenetic status of the established organoids and the initial tumours. 124 In parallel our own group has performed a similar study also based on the differentiation of hPSCs towards exocrine pancreatic cells. Firstly, we generated, efficiently, pancreatic progenitor cells positive for the major pancreatic transcription factors PDX1 and NKX6.1 from cystic fibrosis patients and generated organoids from these progenitors. Subsequently we applied these cultures to a small-scale compound screen for CFTR correctors in 96-well format. Ultimately, two of these compounds clearly influenced forskolin-induced organoid swelling and corrected the observed CFTR-mutant phenotype. 113 This study completes previous investigations on in vitro CFTR disease models focusing on the intestinal phenotype and its correction by genomic editing as well as the nasal mucosa phenotype.86,110,126
Comparably, two studies in CRC have shown that very little tumour tissue as obtained by a fine needle aspiration is sufficient to establish organoids with a success rate of over 70%. 125 Also for the intestinal system the generated organoid cultures fully resemble the genomic and epigenetic state of the initial metastasis they were retrieved from. In line with the preceding study the first HTS (high-throughput screen) in 384-well format was performed on matched organoid pairs from healthy epithelium and adjacent tumours of CRC patients. The organoids were drugged for 6 days by a ‘bespoke 83 compound library’, including clinical drugs as well as chemotherapeutics, drugs from clinical trials and also experimental compounds, followed by cell quantity analysis using CellTiter-Glo. Wisely, the obtained data were correlated to DNA-sequence and RNA-expression analysis and confirmed the mode of action of known compounds but also suggested modes of action for experimental substances. 127
Especially the pioneering work of Hans Clevers and his co-workers on intestinal and now also pancreatic organoids from human biopsy samples paves the road towards personalised medicine. By incorporating the foundation of Hubrecht Organoid Technology (HUB) a milestone is set. All so far collected methodological approaches especially for intestinal organoids will be applied in this foundation. HUB aims for the establishment of Living Organoid Biobanks from comparable healthy and tumorigenic material which can be implemented in high-throughput experimental compound screens in 384-well format, as already published for CRC organoids. 127 The foundation is ‘not-for-profit’ and facilitates licenses to its patented technology to subsequently use this Living Biobank in academic, translational and medical research. 128 Actually the HUB is already far beyond translation to clinics since the foundation signed a license agreement in 2015 with ‘Galapagos’, a clinical stage biotechnology company with its headquarter in Belgium. The agreed aim is ‘the discovery and development of small molecule medicine’ which is already in process with three Phase 2 and two Phase 1 trials, as wells as five pre-clinical studies and 20 discovery programs for small molecules and antibodies on CF, inflammatory, and other diseases. 128
Although organoid screenings are currently a fast-developing field we are not yet at the point of pure instrumentation for therapeutic screenings and translational medicine. Still remaining challenges are the comparability and possible automation of such technologies, the readout of specific organoid systems reflecting different organs and finally the financial reimbursement. Automation and generalisation of organoid systems requires standard operating procedures that can be applied in any laboratory and a ‘common sense’ of sharing protocols. In this context many aspects have to be further specified, e.g. the choice of extracellular matrix for the cells, possible standardisation of size and composition of the organoids, e.g. by 3D-printing approaches, automation of medium formulation, and compound application, e.g. by microfluidic systems. At the moment researchers are searching for applicable and simple readouts like cell proliferation, organoid size and composition, enzymatic activity, organoid swelling that also have to be standardised in context of personalised medicine. Finally, financial aspects have to be considered. Up to now all these studies were developed on basic research funds but, in the near future, with application in personalised medicine, in a variety of different patients, health insurance might have to compensate for the costs of organoid establishment from biopsies and compound testing. This in turn requires standardised, functional and effective protocols and readouts to convince health insurance, e.g. of the forskolin-induced swelling assay as a therapeutic readout. The aspect of costs also supports the combination of organoid screening and microfluidic technology that are minimising the necessary amount of starting material, medium and its components, extracellular matrix, the experimental compounds and finally the chemicals for readout assays. Microfluidic systems hold great potential and could revolutionise the organoid screening technology already proven on hepatic organoids and by high-throughput platform using in vitro stem cell niches.129,130
In summary, high-throughput screening organoid platforms are currently in development for clinical applications but should be further optimised by standardisation of protocols, methods and readouts to realise reliable patient-specific data in all applying disease centres.
Organoid transplantation – a realistic option?
As described above, the group of Hans Clevers demonstrated for the first time complex intestinal organoid formation, initially derived from murine Lgr5+ cells. 72 The potential of iPSC-derived organoids arises from their ability not only to model complex tissue structures ‘in a dish’ but also to fully integrate into intestine structures in vivo. Initial attempts to transplant intestinal organoids (not directly stem cell-derived) have already been performed 10 years ago. 131 Six years later transplantation of organoids that were derived solely from Lgr5+ stem cells were successfully transplanted into superficially damaged murine colon. 132 Donor derived cells formed a layered epithelium exhibiting functional and histological characteristics of normal colon tissue. 132 The next step was the initiation of genetically ‘engineered organoids’, e.g. modulated by CRISPR/Cas9-technology. 86 In turn, such an approach might not only be promising under the aspect of disease modelling but also a potential option of introducing genetically repaired tissue to a place of damage or malfunction within a patient. For pancreatic organoids heterotopic transplantation (kidney capsule transplantation) has already been performed albeit being either restricted to mouse or PSC-derived endocrine organoids without evidence of acinar or ductal components.102,133 In that sense our own group has built up protocols to transplant untransformed pancreatic organoids derived from hPSCs predominantly containing acinar and ductal cells orthotopically into murine pancreas. Indeed, these organoids formed human foetal pancreas without evidence for dysplastic growth. 113 Stable organoids from Lgr5+ liver stem cells turned out to be transplantable in vivo as well. 103 Even the limitation of insufficient vascularisation in transplants could be overcome for iPSC-derived hepatocytes. 134 Thus, not only disease-modelling, but also possible therapeutic applications might arise from stem cell based organoids. Possible applications in CF patients could be terminal liver failure or treatment of pancreoprivic diabetes but also to replace dysfunctional acinar and ductal tissue.
Conclusion
Gastrointestinal (GI) facets of cystic fibrosis (CF) have become more important during the last decade due to an increased life span caused by improved treatment regimen for CF-driven lung disease. In turn, novel treatment and surveillance algorithm for subsequently arsing GI problems such as liver cirrhosis but also GI cancer need to be developed. Also, recent studies provided evidence that treatment success of CFTR correctors and potentiators differs widely across several mutations and individuals.
66
Thus, novel model systems are warranted, which allow drug testing non only in an organ-specific but also more personalised and therefore patient-specific (epi)genetic setting. Here 3D organoids derived from patient-specific induced pluripotent stem cells might be the most promising resource:
They can be derived from diseased individuals non-invasively either from plucked human hair but also from urine samples. Moreover, virtually every organ-phenotype can be mimicked due to the pluripotent nature of these cells. The latter characteristic also allows indefinite replenishment for biochemical assays. Also the co-differentiating niche cells arising from PSCs allow cost-effective culture and growth without the requirement of many expensive growth factors. Finally, developmental conclusions can be drawn and large-scale, tailored and individualised drug screens become possible.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was funded by the Deutsche Forschungsgemeinschaft (DFG, K.L. 2544/1-1 and 1-2), the Forschungskern SyStaR to AK, BIU (Böhringer Ingelheim Ulm to AK), the Fritz-Thyssen Foundation (Az. 10.15.2.040), the German Cancer Aid (111879) and the Else-Kröner-Fresenius-Stiftung (2011_A200). AK is indebted to the Baden-Württemberg Stiftung for the financial support of this research project by the Eliteprogramme for Postdocs. AK is also an Else-Kröner-Fresenius Memorial Fellow. LP is supported by a research fellowship of the Else-Kröner-Fresenius- Stiftung. MH was supported by the International Graduate School in Molecular Medicine and the Bausteinprogramme (L.SBN. 110), Ulm University. MM is supported by a grant of Ulm University (Baustein for Senior Clinician Scientists).