
Abstract
Background
Irritable bowel syndrome (IBS) is a common functional gastrointestinal disorder characterized by recurrent abdominal pain and/or discomfort. Probiotics have been reported to benefit IBS symptoms but the level of benefit remains quite unclear.
Objective
This study was designed to assess the benefit of Saccharomyces cerevisiae I-3856 on IBS symptoms.
Methods
A randomized, double blind, placebo-controlled trial has been performed in 379 subjects with diagnosed IBS. Subjects were randomly supplemented with the probiotics (1000 mg) or placebo for 12 weeks. Questionnaires (gastrointestinal symptoms, stools, wellbeing, and quality of life) were completed. Primary endpoint was percentage of responders defined as having a 50% decrease in the weekly average “intestinal pain/discomfort score” for at least 4 out of the last 8 weeks of the study.
Results
There was no overall benefit of S. cerevisiae I-3856 on IBS symptoms and wellbeing in the study population. Moreover, S. cerevisiae I-3856 was not statistically significant predictor of the responder status of the subjects (p > 0.05). Planned subgroup analyses showed significant effect in the IBS-C subjects: improvement of gastrointestinal symptoms was significantly higher in active group, compared to placebo, on abdominal pain/discomfort and bloating throughout the study and at the end of the supplementation.
Conclusions
In this study, S. cerevisiae I-3856 at the dose of 1000 mg per day does not improve intestinal pain and discomfort in general IBS patients. However, it seems to have an effect in the subgroup with constipation which needs further studies to confirm (NCT01613456 in ClinicalTrials.gov registry).
Introduction
Irritable bowel syndrome (IBS) is a ubiquitous functional gastrointestinal disorder characterized by abdominal pain or discomfort, associated with altered bowel habits, 1 reported by 10% of the general population with a female predominance. 2 It has negative impact on health-related quality of life and induces high health costs. Despite increased understanding there have been few new therapies introduced, and a combination of rare adverse effects and very low tolerance of side effects has led to the withdrawal of some effective therapies, notably alosetron and tegaserod.
Recent interest has therefore focused on drugs (e.g. lubiprostone, linaclotide) or agents like pro- and prebiotics whose actions are confined to the lumen with a perceived low likelihood of unwanted systemic effects.
There are numerous studies suggesting possible benefit of probiotics on IBS symptoms but the results are variable.3–8 This heterogeneity arises because different strains have different modes of action and also because IBS patients were often unselected and hence heterogeneous as regards bowel habit and underlying disease mechanism. Regrettably, few studies have included a mechanistic component so it is unclear exactly which of their numerous putative effects demonstrated in the laboratory are responsible for benefit in clinical practice.
Saccharomyces cerevisiae I-3856, a strain from Lesaffre baker’s yeast collection, in common with most yeasts secretes numerous saccharolytic enzymes which may assist the intestinal microbiota in generating short chain fatty acids (SCFAs) and alcohols which are known to exert a prokinetic effect at least in the small intestine. 9 This suggests that, like Bifidobacterium lactis, 10 S. cerevisiae I-3856 might be most effective for IBS with constipation. Furthermore, it has already demonstrated a strong visceral analgesic effect and decreased perception of pain in a rodent model of colonic hypersensitivity. 11 This property also suggested that it might benefit all subtypes of IBS given that visceral hypersensitivity is thought to be important in many IBS patients. 12 Preliminary data from the first randomized placebo-controlled trial (RCT) of S. cerevisiae I-3856 at the dose of 500 mg daily, in 179 unselected IBS patients, 13 showed a significantly higher proportion of responders achieving a 50% reduction in average pain score in the active group compared to the placebo group in the last 4 weeks of treatment.
Based on these preliminary data, we aimed to confirm these findings in another RCT using a larger dose of S. cerevisiae I-3856 (1000 mg daily) and greater numbers of IBS subjects who were carefully phenotyped as to bowel habits.
Materials and methods
We performed a multi-center, randomized, double blind, placebo-controlled trial according to Good Clinical Practice Guidelines, Declaration of Helsinki, and approved by the Ethics Committee Ouest IV of Nantes (France). Signed written informed consent for the study was obtained from all subjects before protocol-specific procedures were carried out and subjects were informed of their right to withdraw from the study at any time.
Patients
Men and women (18–75 years of age) were recruited by French general practitioners (GPs) and in a clinical investigation center (Nantes, France). Patients were required to be diagnosed with IBS by their GP and to meet the Rome III criteria, 1 with pain or discomfort present >1 day per week; and a score for abdominal pain or discomfort >1 and <6, determined at the beginning of the study on a seven-point Likert scale for pain/discomfort scored from 0 (no pain or discomfort) to 7 (severe pain or discomfort). Lactose intolerance was excluded in subjects characterized by IBS with diarrhea (IBS-D) and mixed IBS (IBS-M) by means of a milk challenge (no symptoms after consuming 500 mL low fat milk after an overnight fast). All patients had normal full blood count and routine biochemical screen, transglutaminases IgA, fecal calprotectin, and C-reactive protein (CRP) levels. Long-standing treatments for diarrhea, laxatives, and antispasmodic drugs were not an exclusion criterion provided the doses were stable and not modified during the study. Menopausal females receiving a hormone treatment or contraception in non-menopausal female subjects must have started their treatment at least 3 months beforehand and remained on a stable dose for the entire duration of the study.
Subjects were excluded if they had chronic gastrointestinal disorders, history of gluten intolerance (celiac disease), or elevated tissue transglutaminase IgA titers, treatments likely to influence IBS in particular by modifying intestinal sensitivity or motility (antidepressants, opioids, and narcotic analgesics), antibiotic therapy in progress or prescribed in the 8 weeks before inclusion in the study, long-term treatment with analgesics or non-steroidal anti-inflammatory drugs. Subjects not willing to stop taking probiotics, prebiotics, or synbiotics in the form of dietary supplements or convenience goods were not eligible. Pregnancy, chronic alcoholism, vegetarian or vegan subjects, eating disorders such as anorexia or bulimia, and documented food allergies were also exclusion criteria.
Study design
The 12-week study included 364 subjects divided in two parallel groups. Subjects visited the clinical center at four visits (Figure 1): V0 (pre-inclusion visit), V1 (inclusion visit, allocation of the study products), V2 (4 weeks after V1, follow-up visit) and V3 (12 weeks after V1, end-of-study visit).
Study design.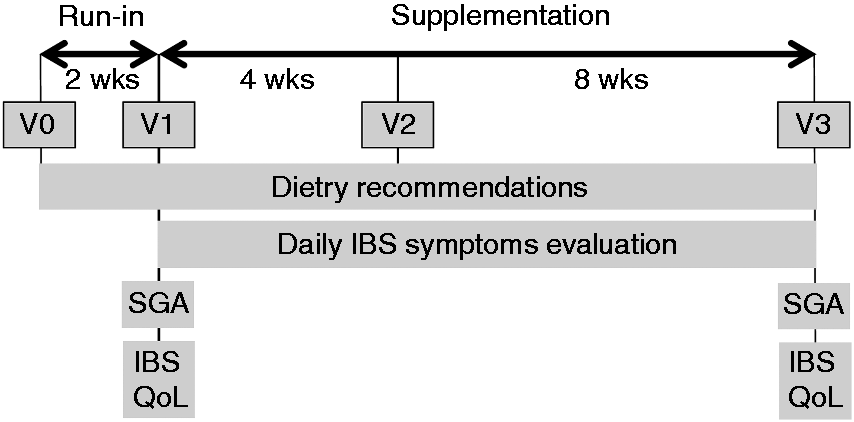
Pre-inclusion visits were planned to check inclusion and exclusion criteria and perform screening tests. Bristol stool scale (BSS) questionnaire and IBS symptoms, including abdominal pain/discomfort, were evaluated daily for a two-week period. Additionally, dietary recommendations were explained to the subject, in order to avoid eating any foods containing prebiotics and/or probiotics and any fermented milk products. At inclusion visit, investigators enrolled and randomized eligible IBS patients to consume either S. cerevisiae I-3856 (1000 mg, 8 × 109 colony forming units (CFU)/g) (active group) or a placebo (calcium phosphate and maltodextrin) (placebo group), daily, for 12 weeks. Randomization was done with a block method and without stratification, using SAS® software version 9.1.3 Service Pack 4 (SAS Institute Inc., Cary, NC, USA). It was drawn up before the beginning of the study by a person not related to the clinical phase, the data management, or statistics. A randomization management interface was used by the investigators to allocate randomization numbers to included subjects. The randomization list which links each subject to a product group was prepared and stored confidentially. The unblinding envelopes were concealed from the person responsible for randomization. From V1 to V3, patients had to complete a daily diary documenting the frequency and consistency of their stools (BSS), 14 and the intensity of IBS symptoms (abdominal pain/discomfort, bloating, flatulence/borborygmi, and difficulty with defecation) on Likert scales rated from 0 (null) to 7 (severe). 15 They also had to answer weekly some questions related to their bowel movements, in order to assess the number of complete spontaneous bowel movements. Subjective global assessment (SGA) was assessed once a week from their answer to the following question: “Have you had satisfactory relief from your IBS symptoms in the last 7 days?” (binary endpoint Yes/No). 16 Finally, they completed an IBS-specific quality of life questionnaire (IBS QoL) at V1 and V3. 17
Study products and compliance evaluation
Study products were presented in capsule form, packaged in plastic bottles. Active and placebo products were similar in color, form, flavor, and size. During the whole study neither the investigators nor the subjects were aware of the product tested. Subjects were instructed to consume orally two capsules of 500 mg per day in the morning, during breakfast, together with a glass of water.
The I-3856 strain is a proprietary, well characterized strain of Lesaffre, registered in the French National Collection of Cultures of Microorganisms (CNCM). The S. cerevisiae species is characterized by using phenotypic (API® ID32C, Biomerieux SAS) and genotypic referenced methods (genetic amplification and sequencing of 26S DNA).18,19 Moreover, the strain I-3856 is identified thanks to the polymerase chain reaction (PCR) Interdelta typing technique, 20 and other genetic methods (e.g. complete genome sequencing).
Patients had to return all their capsule bottles, containing unconsumed capsules at V3. Compliance was calculated during the treatment period at visit V3, by comparison of the theoretical number of capsules consumed during the individual supplementation period and the number of capsules returned.
Study endpoints
The primary endpoint defined in the protocol was the percentage of responders, i.e. the percentage of patients who had an improvement of 50% of the weekly average “intestinal pain/discomfort score” compared with baseline average score (V0-V0bis) for at least 4 out of the last 8 weeks of the study (V2 to V3). This was based on previous experience suggesting S. cerevisiae I-3856 could increase the proportion of IBS patients meeting this demanding end point. Secondary endpoints were IBS symptoms scores (individual scores and composite score (defined as the sum of the four individual scores)), stools characteristics in IBS types, quality of life, and weekly SGA. 13
Safety endpoints
Safety of the study was assessed considering the occurrence of adverse events, safety blood parameters, and the follow-up of vital signs (blood pressure and heart rate). Adverse events were recorded throughout the study and investigators had to estimate their severity and to judge whether they were related to treatment.
Statistical considerations
Sample size was calculated to be 172 subjects per arm in order to detect a difference of 15% between active and placebo groups for the primary criteria, assuming a 47% placebo response rate, 13 using a bilateral binomial test with 80% power and alpha risk of 5%. Allowing for a 5% of drop-out, we planned to enroll a total of 364 subjects.
Data were analyzed using SAS® software version 9.3 (SAS Institute Inc., Cary, NC, USA). Results are expressed as Mean ± SD or Estimated Mean ± SE. Significance was set at p < 0.05.
Primary endpoint (percentage of responder) was analyzed using a logistic regression model. IBS symptoms and data from BSS were analyzed applying a mixed model Analysis of Covariance (ANCOVA) for repeated measurements, SGA was analyzed applying a generalized estimating equation model for correlated binary variable, and quality of life scores were analyzed using analysis of variance (ANOVA).
Primary analysis was performed on the full set of patients, in both intent-to-treat (ITT) and per protocol (PP) populations. Additional analyses were performed in subsets of IBS population (IBS-C, IBS-D, and IBS-M), based on Rome III definition. 1 Individual IBS symptoms and composite score were analyzed weekly and as area under the curve (AUC) (calculated on the 12 weeks of the study).
Results
Subjects
Baseline characteristics of subjects
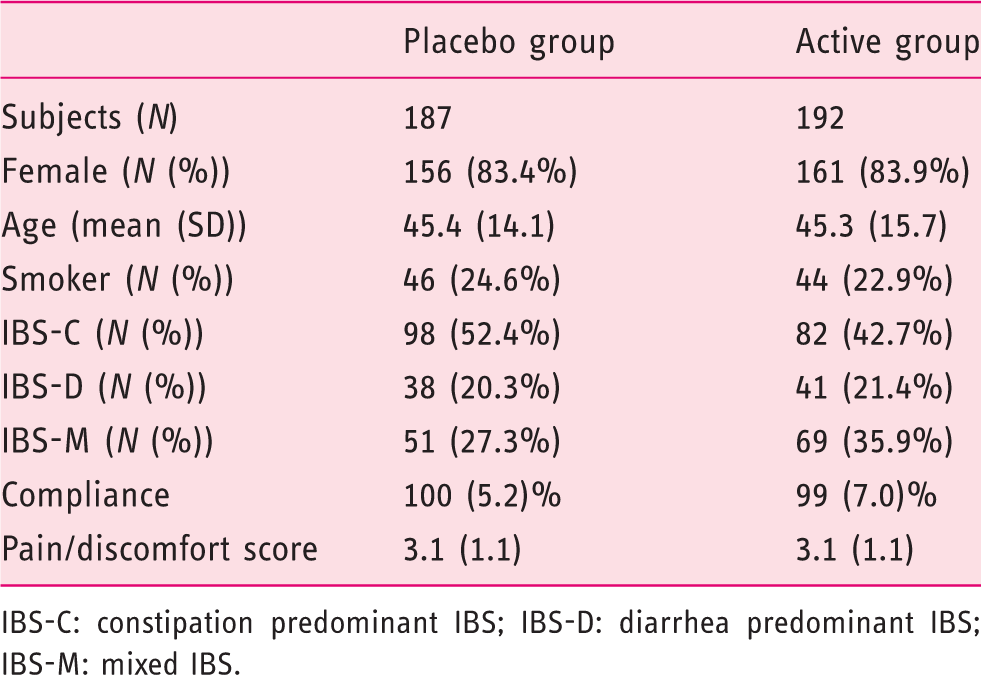
IBS-C: constipation predominant IBS; IBS-D: diarrhea predominant IBS; IBS-M: mixed IBS.
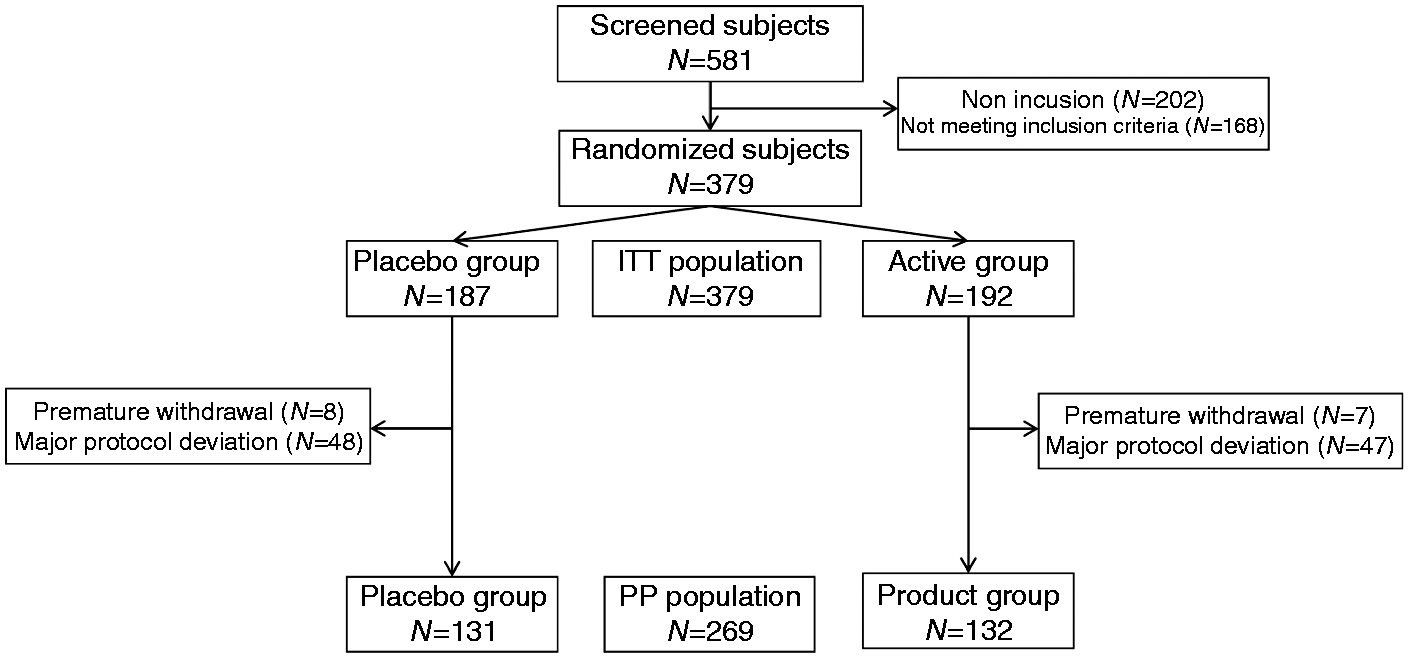
Flow chart of subjects throughout the study.
Fifteen (15) subjects did not complete the study (protocol deviation (N = 10), subject not willing to continue the study (N = 3), worsening of symptoms (N = 1), pregnancy (N = 1)) leaving, respectively, 185 and 179 subjects in the active and placebo groups at the end of the study. A majority of subjects (47%) were IBS-C subjects (respectively, 43% and 52% in the active and placebo groups). Good compliances were recorded in the active and placebo groups throughout the 12 weeks of supplementation (respectively, 100 (5.2)% and 99 (7.0)%). The ITT population was defined as all subjects who were randomized (N = 379, active group N = 192, placebo group N = 187) whereas PP population included subjects of the ITT population presenting no major protocol deviations, and where data on primary endpoint were available for at least 7 weeks out of the last 8 weeks of the study (N = 269, active group N = 138, placebo group N = 131). The main reasons for exclusion of subjects from the PP populations are deviations on inclusion/exclusion criteria (N = 33 subjects), consumption of prohibited concomitant medications during the study (N = 46), deviations on visit dates (N = 31), and consumption of probiotics during the study (N = 11). Analysis of the PP population did not show any difference from the ITT population so all the data shown below are on an ITT basis.
Results on total population
The responder status of subjects was analyzed during the last 8 weeks of the study in subjects without missing data. Forty-seven (47) subjects out of 175 in the placebo group (26.9%) and 57 subjects out of 177 in the active group (32.2%) were considered as responders, which means that they have an improvement of 50% of the weekly average “intestinal pain/discomfort score” compared with baseline average score for at least 4 out of the last 8 weeks of the study. However, the active product was not a statistically significant predictor of the responder status (p > 0.05). Considering abdominal pain/discomfort score, significant decrease was observed from the beginning to the end of supplementation, in both active and placebo groups (respectively, −0.61 and −0.43, p < 0.0001 in both groups). No statistically significant product effect adjusted on baseline and IBS type was noted all weeks taken together. Similar observations were reported on bloating, flatulence/borborygmi, difficulty with defecation, and composite score of IBS symptoms, with statistically significant improvements during supplementation in the two groups.
A relief in IBS symptoms was reported by subjects as soon as the first week of supplementation (27.2% of subjects of the placebo group, 33.6% in the active group), with statistically significant improvement at the end of supplementation (57.7% of subjects of the placebo group, 60.9% in the active group at the last week of supplementation, p < 0.0001 in both groups). Finally, quality of life was significantly improved in placebo and active groups throughout the study (p < 0.0001), with no statistically significant difference between groups.
Subgroup post hoc analysis
Baseline characteristics of subjects in the subgroups of IBS
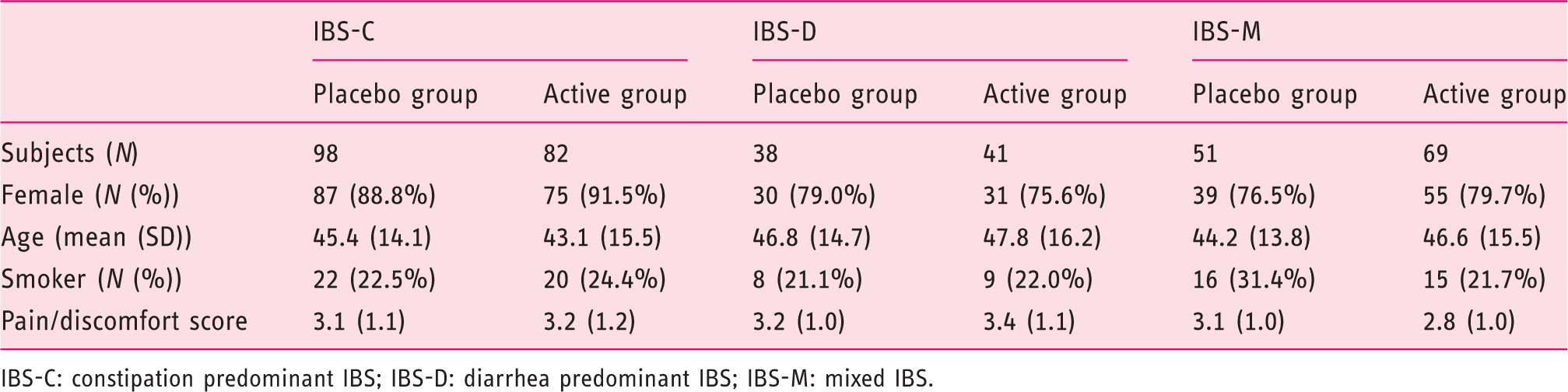
IBS-C: constipation predominant IBS; IBS-D: diarrhea predominant IBS; IBS-M: mixed IBS.
IBS symptoms scores (mean (SD)) during the study in placebo and active IBS subgroups, before (W0) and after (W12) supplementation
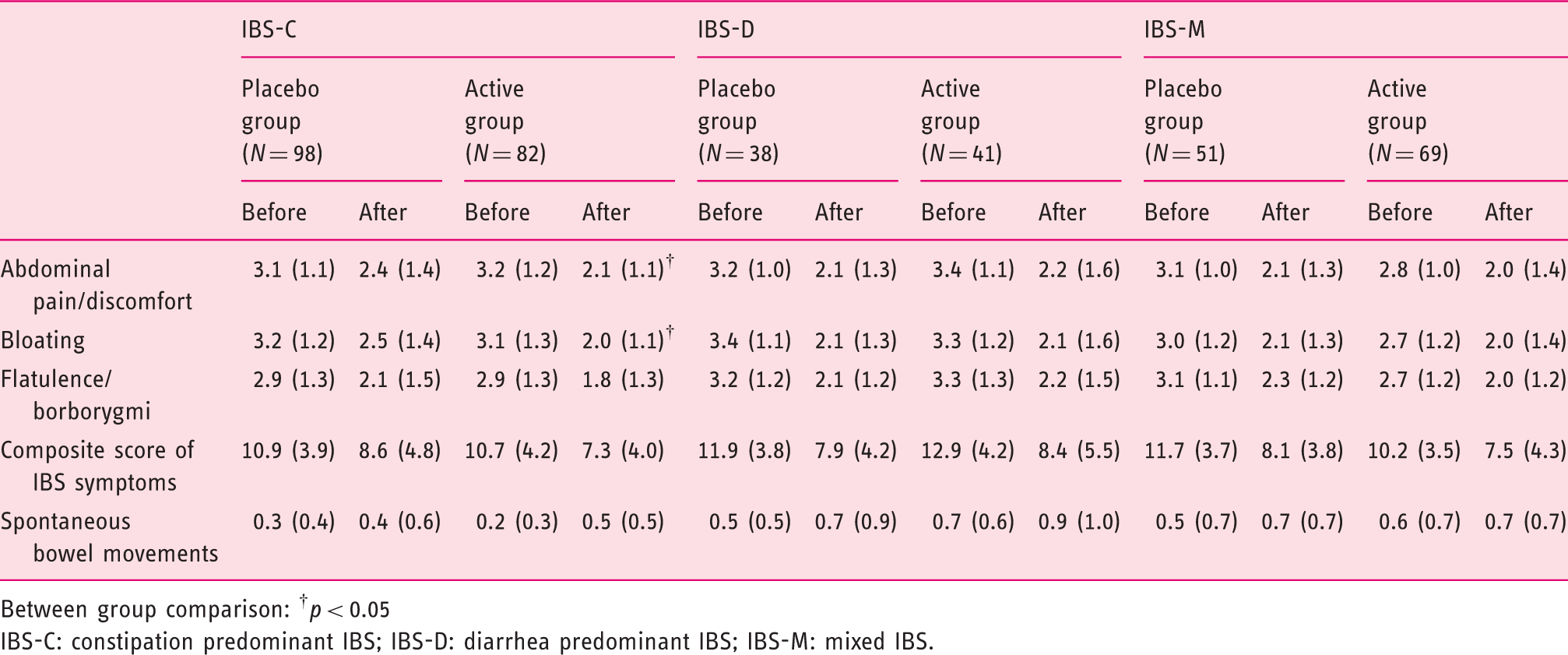
Between group comparison: †p < 0.05
IBS-C: constipation predominant IBS; IBS-D: diarrhea predominant IBS; IBS-M: mixed IBS.
At the end of the supplementation period, significant improvement was observed in abdominal pain/discomfort, bloating, flatulence/borborygmi, and composite score in both active and placebo groups of IBS-C subjects (Table 3) but greater on active than placebo for abdominal pain/discomfort (p = 0.03) and bloating (p = 0.03). A similar trend was observed with the composite score of IBS symptoms (7.3 (4.0) in the active group vs. 8.6 (4.8) in the placebo group) but this just failed to reach statistical significance (p = 0.05) (Figure 3(a) to (c)).
Evolution of abdominal pain/discomfort (a), bloating (b) and composite score of IBS symptoms (c) in IBS-C subjects, during the 12-week consumption of active product (S. cerevisiae I-3856; crosses and discontinued line) and placebo product (circle and black line). Each IBS symptom was rated from 0 (null) to 7 (severe); composite score corresponds to the sum of the abdominal pain/discomfort score, bloating score, flatulence/borborygmi score, and difficulty with defecation score. Values are estimated means, with standard error represented by vertical bars. Statistically significant differences between groups are symbolized by *(p < 0.05).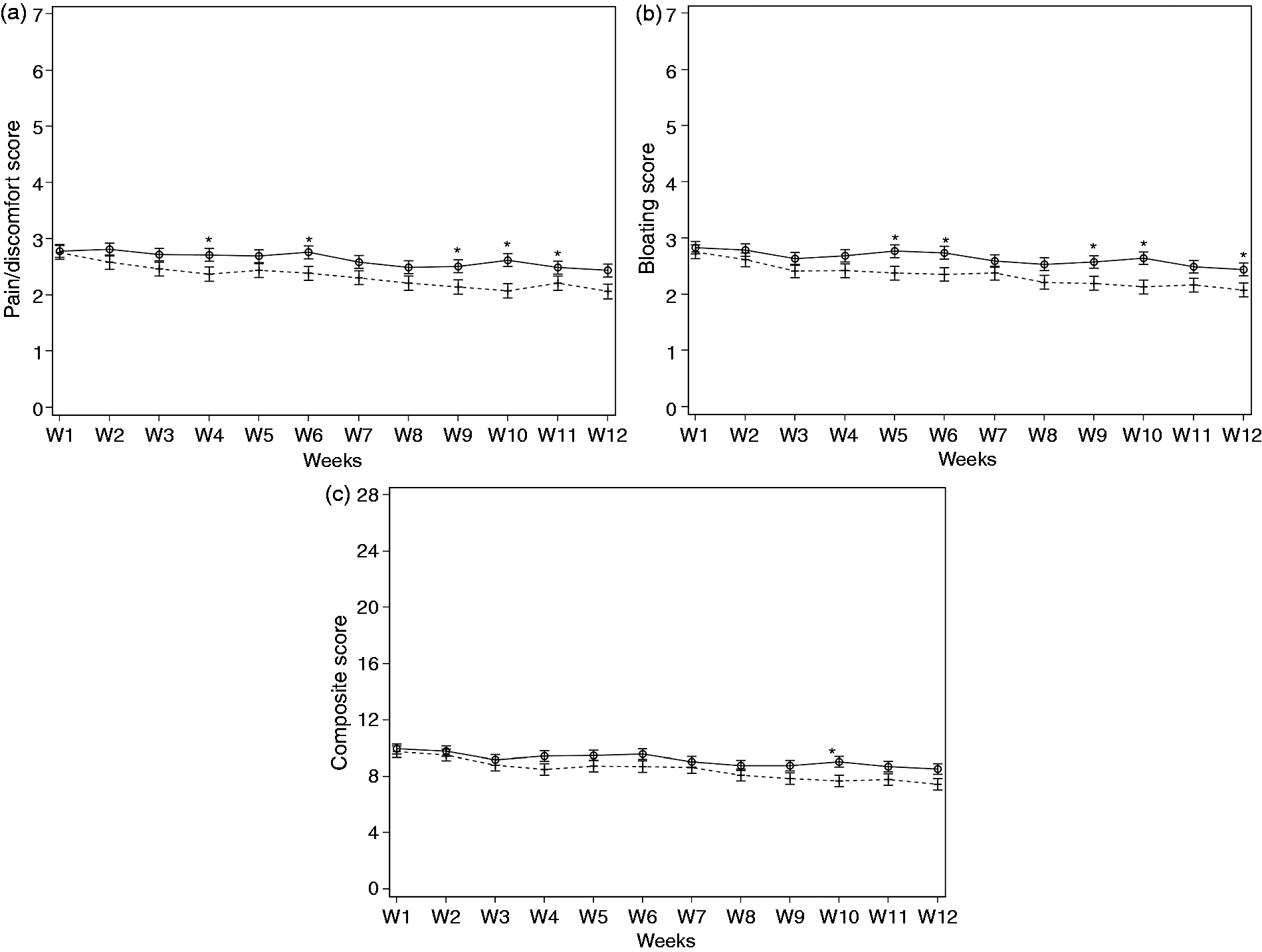
In IBS-C subgroup, the beneficial effect of S. cerevisiae I-3856 is also observed throughout the study, with significantly lower total AUC for abdominal pain/discomfort (−10.9%, diff [CI95%] = −3.56 [−6.99; −0.13], p = 0.04) and bloating (−13.6%, diff [CI95%] = −4.44 [−7.71; −1.18], p = 0.01) in the active group, compared to placebo group (Figure 4(a) to (c)).
AUC of abdominal pain/discomfort (a), bloating (b) and composite score of IBS symptoms (c) in IBS-C subjects over the 12 weeks of active product (S. cerevisiae I-3856; black bar) and placebo product (white bar) consumption. The AUC of the composite score corresponds to the sum of the AUC of the four IBS symptoms: abdominal pain/discomfort, bloating, flatulence/borborygmi, and difficulty with defecation Values are estimated means, with standard error represented by vertical bars. Statistically significant differences between groups are symbolized by *(p < 0.05) and **(p < 0.01).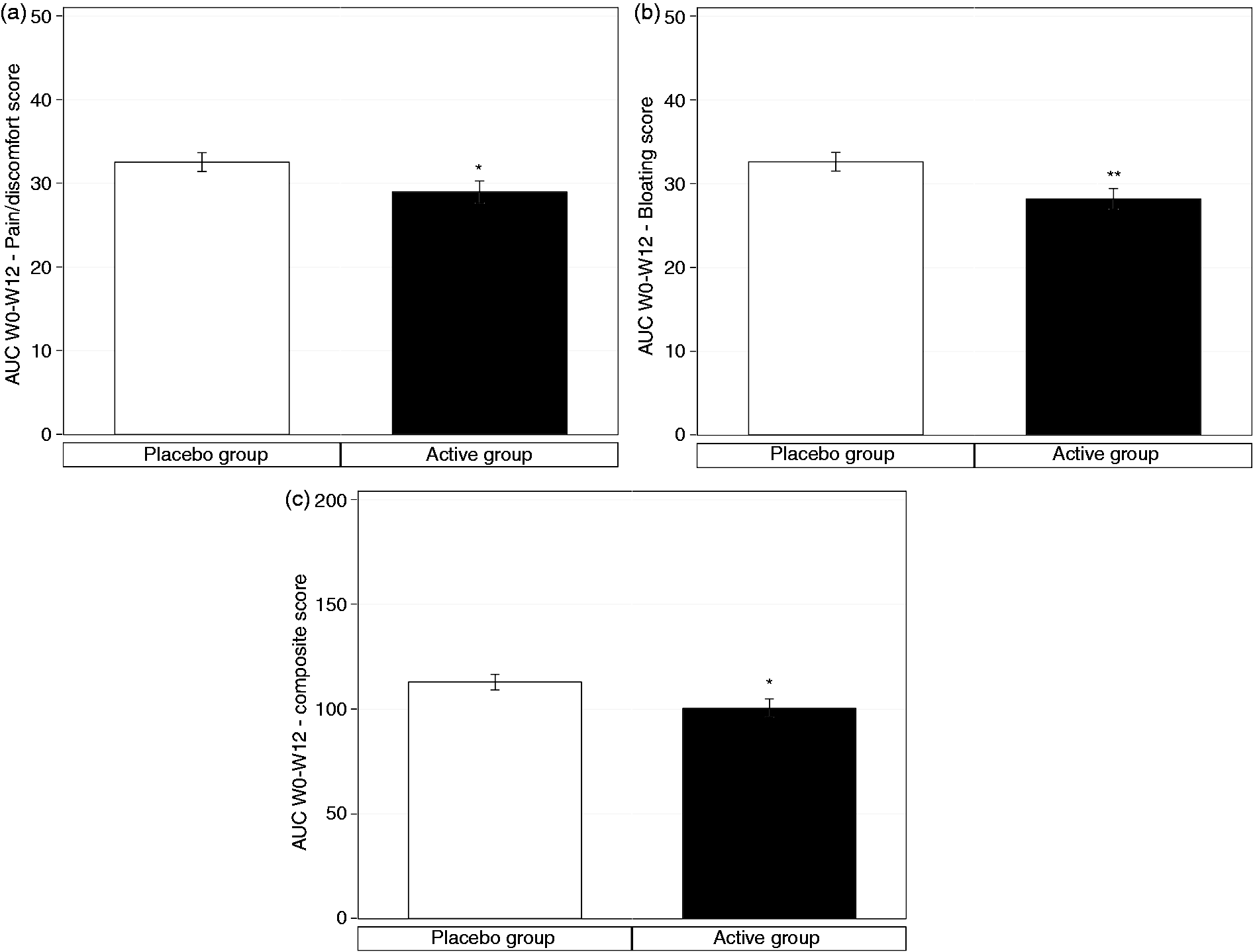
The composite score of IBS symptoms similarly demonstrated significantly lower total AUC in the active group, in comparison to the placebo group (−11.0%, diff [CI95%] = −12.46 [−23.76; −1.16], p = 0.03). Total AUC of quality of life was positively correlated to the AUC of pain/discomfort (r = −0.28887, p = 0.05). There was, however, no statistical difference in the proportion of responders in the active (22.0%) and in the placebo group (21.4%). The number of complete spontaneous bowel movements showed numerically higher value on active group at each week of supplementation period but this did not reach statistical significance. However, analysis of covariance of the weekly average BSS score (stool consistency) showed a statistically significant interaction between tested product and time (p = 0.04): post hoc analysis showed a significant difference only at week 8 with average BSS score of median (IQR) 3.0 (2.0–3.9) on active versus 2.2 (1.7–3.4) on placebo (p = 0.00), diff [CI95%] = 0.447 [0.147; 0.746].
Safety
No serious adverse event linked to the research or to the study product was recorded during the study in the placebo and product groups. Sixteen adverse events were judged as possibly linked to the research or to the study product by the investigators, and among these 16 adverse events, 14 were gastrointestinal (constipation (4 in active group, 2 in placebo group), abdominal pain (2 in active group), gastroesophageal reflux (1 in active group, 1 in placebo group), etc.): 10 were reported in the active group and 4 in the placebo group.
Discussion
The results of this randomized, placebo-controlled clinical study demonstrate that S. cerevisiae I-3856 at the dose of 1000 mg daily does not alleviate gastrointestinal symptoms in unselected IBS patients. The choice of the somewhat demanding primary endpoint of this study, namely 50% reduction in average pain score, was based on previous RCT, 13 which demonstrated positive effect on abdominal pain/discomfort. The sample size determination was calculated on the basis of this previous clinical study. We hypothesized a difference of 15% between the two groups. However, this hypothesis may be too optimistic making our study underpowered to detect the small difference observed. In the present RCT, around half our patients (N = 180) had IBS-C and this subgroup does appear to benefit with notable reduction in abdominal pain/discomfort, bloating and composite scores of IBS symptoms. Pain scores fell 1.1 on a seven-point scale, which is similar to the fall observed in other recent trials of IBS-C,21,22 representing a 34% reduction from baseline. This current larger study was designed to confirm pilot data observed with the same yeast strain in unselected IBS patients where a greater improvement in pain was noted with a single 500 mg daily dose, given for 8 weeks. 13 The current study used a higher dose (1000 mg daily) which was well tolerated. The larger number and more detailed characterization of the IBS subtype allowed us to see that the beneficial effect was confined to those with constipation. A previous mechanistic study in IBS with constipation using Bifidobacterium infantis showed an acceleration of both small and large bowel transit. 10 Accelerating small bowel transit would be predicted to increase the influx of ileal contents into the caecum and secondarily accelerate colonic transit. 23 The underlying mechanism related to accelerated bowel transit is unclear but could relate to the known stimulatory effect of SCFAs on ileal motility. 9 These are likely to act via fatty acid receptors on enteroendocrine cells. S. cerevisiae yeasts have extensive fermentative capacity and may increase small bowel SCFA levels though this has yet to be confirmed experimentally. It is worth noting that the presence of large numbers of live organisms in the upper small bowel is the major change induced by consuming probiotics, given the low level of organisms normally present. In contrast, in the colon, the impact is much less as they are diluted by the 1013 organisms already present. This can be seen more clearly now that we can accurately non-invasively measure gut volumes using Magnetic Resonance Imaging. The normal fasting small bowel volume averages just 90 mL, 24 while the fasting colon volume averages 561 mL. 25 The commensal bacterial titer is low, usually below 105 while the titer of the colon can reach a concentration of 1013/mL of normal commensals. Consequently, assuming a uniform distribution, the ingestion of two capsules of S. cerevisiae I-3856 containing 8 × 109 CFU/g would barely alter colonic organism numbers while increasing small bowel organism numbers 40,000 fold. In our study, the hypothesis of accelerated transit has not been validated by measure of the colonic transit time. However the number of complete spontaneous bowel movements reported by subjects was numerically higher on active group in IBS-C subtype and the stools did tend to be softer on active group compared to placebo, suggesting transit may well have been accelerated. Other studies measuring transit before and after treatment would be required to test the hypothesis of accelerated transit. Subjects included in the present protocol were not tested for small intestinal bacterial overgrowth (SIBO), which has been reported in 4–19% of IBS patients,26,27 the precise incidence depending on the methods used. Its role in causing IBS symptoms is contentious but some studies suggest a modest benefit from antibiotic use. 28 At present it is not usually used in screening patients subjected to clinical trials but if a more reliable method of testing was available this might be reconsidered in the future, especially when testing products like probiotics which might affect gut bacteria. Another consideration is whether a low FODMAP diet, as advocated by the group from Monash University to treat IBS patients complaining of bloating and flatulence, 29 should be tried before entering patients into future clinical trials. At present this is not standard practice but if more evidence from other units support this then perhaps this should change.
Why S. cerevisiae benefits pain/discomfort remains quite unclear. While animal studies suggest it might reduce visceral hypersensitivity, the translation of such studies to benefit in clinical studies has a poor track record, 17 so the relevance of the animal studies is quite unclear. Recent evidence supports the idea that pain in IBS-C increases as the number of days without bowel movements increases. This is possibly due to increasing pressure being required to propel the harder stool that results from slow transit. The increases in Bristol Stool Scale score we observed which make the stool softer may therefore account for the improvement in pain. Acceleration of transit in both small and large bowel has been linked to reduction in bloating, 10 so this is a possible mechanism which could be tested in future studies.
Our results provide additional data to those obtained before on the beneficial effect of S. cerevisiae I-3856 in IBS. 6 Although there was no effect of S. cerevisiae supplementation on IBS symptoms and quality of life in the total population, S. cerevisiae I-3856 improved gastrointestinal symptoms in the subgroup with constipation. Future studies should examine the underlying mechanisms, particularly the changes in small bowel transit to enable better targeting of treatment to patients who will benefit.
Footnotes
Acknowledgments
All authors have approved the final version of the article and the list of authors. The authors would like to thank subjects for their interest, time and compliance with the supplementation. Writing support was provided by Gunnard K Jacobson of Lesaffre Yeast Corporation.
Funding
This work was realized in the frame of the collaborative research program LEVACI supported by the French FUI fund (Fonds Unique Interministériel), the FEDER (European Fund for Regional Development), the Region Nord-Pas-de-Calais and LMCU (Lille Métrople Communauté Urbaine) in France. LEVACI was approved by the French competitiveness clusters “Nutrition-Health-Longevity” and “Vegepolys”. The writing of this paper was funded by Lesaffre.
Conflict of interest
Prof. Robin Spiller has served as an expert for Lesaffre and Biofortis and has received consulting fees and research funding from Lesaffre.
Fanny Pelerin, Amelie Cayzeele Decherf, and Dr Peter Jüsten are employees of Lesaffre.
Corinne Maudet, Béatrice Housez, and Murielle Cazaubiel are employees of Biofortis. Biofortis has received funding from Lesaffre.
Notes
The study was registered on ClinicalTrials.gov registry under identifier NCT01613456. NB: At the time of the study registration, an error was made stating that the primary endpoint was evaluated over 12 weeks when in fact the protocol specified an evaluation over the last 8 weeks of treatment. Unfortunately, this point was only rectified on clinicaltrials.gov after study completion. However, the study published in this Journal was carried out exactly according to the approved protocol and the primary endpoint was never changed, neither before nor after the trial was started.