
Abstract
Inflammatory bowel disease represents a chronic intestinal inflammation. Recent knowledge suggests a crucial role for genetic, immunological and bacterial factors in inflammatory bowel disease pathogenesis. Variations within the gene locus encoding PTPN22 have been associated with inflammatory bowel disease. PTPN22 is critically involved in controlling immune cell activation and thereby plays an important role in maintaining intestinal homeostasis. Although in B and T cells the mechanism showing how PTPN22 affects cell signalling pathways is well studied, its role in myeloid cells remains less defined. Regulation of the innate immune system plays an essential role in the intestine, and levels of PTPN22 in myeloid cells are drastically reduced in the intestine of inflammatory bowel disease patients. Therefore, additional studies to define the role of PTPN22 in myeloid cells might clearly enhance our understanding of how PTPN22 contributes to intestinal homeostasis.
Keywords
Introduction
Inflammatory bowel disease (IBD) is comprised of two major sub-forms, called Crohn’s disease (CD) and ulcerative colitis (UC). While UC is a continuous inflammation restricted to the colon, CD is a discontinuous inflammation that can affect the whole gastrointestinal tract. Apart from chronic and relapsing intestinal inflammation, a characteristic feature of UC and CD is the presence of extra-intestinal manifestations, such as inflammation in the joints, eyes or the skin. 1
According to current knowledge, genetic, immunological and bacterial factors contribute essentially to the pathogenesis of IBD. 2 A dysfunctional immune response of the innate as well as the acquired immune system to commensal microbes results in either excessive up- or impaired down-regulation of inflammatory events, which finally drive the development of chronic intestinal inflammation.2,3 The predispositions for these aberrant immune responses are genetically determined and genome wide association studies (GWAS) identified single nucleotide polymorphisms (SNPs) in more than 160 gene loci being associated with IBD. 4 Many of the identified risk genes are critically involved in controlling host immune responses to the intestinal microbiota – that is, they mediate bacterial recognition, 5 induction of antimicrobial factors or activation and modulation of innate as well as adaptive immune responses.5,6
Phosphorylation and dephosphorylation represent a fundamental mechanism for the activation and inactivation of intracellular signalling molecules. 7 Especially in immune cells, phosphorylation/dephosphorylation of signalling molecules is crucial for an adequate response towards extracellular stimuli such as presence of antigens or cytokines.8–12 While phosphorylation is mediated by protein kinases, dephosphorylation is carried out by a large number of different protein phosphatases. 13 One important family of phosphatases are protein tyrosine phosphatases (PTPs). Members of this family play an essential role in the regulation of critical cell signalling events – that is, proliferation, differentiation and cell survival. 14
In the past decade, GWAS revealed a strong association between SNPs in the gene locus encoding one of these PTPs, namely protein tyrosine phosphatase non-receptor type 22 (PTPN22), and several inflammatory disorders, including CD,4,15 UC,4,15 systemic lupus erythematodes (SLE), 16 rheumatoid arthritis (RA),17,18 type 1 diabetes (T1D) 19 and many others. 20 Nowadays, genetic variants in the PTPN22 locus are considered one of the most important, non-HLA-associated genetic risk factors for the onset of autoimmune disorders, 21 which highlights the importance of PTPN22 in regulating immune responses. In this review we summarize the most recent knowledge about the role of PTPN22 in the pathogenesis of chronic intestinal inflammation, in particular IBD.
Variants in the gene locus encoding PTPN22
Two SNPs within the gene locus encoding PTPN22 are associated with inflammatory disorders, namely SNP rs2476601 (C1858 > T) and SNP rs33996649 (G788 > C). Presence of rs2476601 results in the substitution of arginine 620 with a tryptophan residue in the protein product (referred to as 620W variant). SNP rs33996649 mediates substitution of arginine 263 with a glutamine residue (referred to as 263Q variant).
Arginine 263 lies within the catalytic domain of the PTPN22 protein product and substitution with a glutamine residue results in a decreased phosphatase activity. 22 Of note, presence of the 263Q variant reduces the risk to develop SLE and RA22,23 and protects from UC onset, 15 however not much is known about the effects of this variant on cellular mechanisms and immune functions. For this reason, this variant will not be discussed in more detail in this review.
Presence of the PTPN22 620W variant is associated with an increased risk for RA,17,18 SLE 16 and T1D, 19 but interestingly reduces the risk of developing CD.15,24 The PTPN22 protein has three domains: an N-terminal catalytic domain, followed by a linker domain, and a C-terminal Proline rich domain. 25 Arginine 620 lies within the C-terminal Proline rich domain that seems to be important for determining substrate specificity. 26 Although the 620W variant has initially been described to result in a true gain-of-function protein product, 27 the functional consequence of this substitution has recently been debated. Initial studies by Vang et al. demonstrated a gain-of-function in inhibition of the T cell receptor (TCR) signalling cascade resulting from the 620W substitution, 27 and this variant features increased in vitro de-phosphorylation capacity. 27 The enhanced phosphatase activity might result from reduced PTPN22 phosphorylation at an inhibitory residue.28,29 Other mechanism to explain the enhanced phosphatase activity of the PTPN22 620W variant, include increased substrate interaction due to enhanced localization in lipid rafts, 26 and/or reduced interaction with Csk.30,31 Current hypotheses suggest that in resting T cell Csk binds to PTPN22 and sequesters it away from its substrates in the TCR signalling cascade. 26 620W PTPN22, however, seems to be unable to interact with Csk, resulting in increased interaction of PTPN22 with its substrates, finally resulting in gain-of-function inhibition of TCR signalling. 26 In contrast to the gain-of-function hypothesis, one study using mice that expresses PTPN22 619W (the murine orthologue of the disease associated human PTPN22 620W) indicates that this variant might result in reduced protein stability and therefore finally in a net loss-of-function effect. 32 However, this finding could not be confirmed in other studies using an independently generated PTPN22 619W knock-in strain. 33 Interestingly, in both studies using PTPN22 619W knock-in mice, presence of the PTPN22 619W variant resulted in increased T cell responses, 33 an effect that contradicts the gain-of-function hypothesis.
In an approach using in vivo silencing of PTPN22 by siRNA interference, phenotypically opposite effects of the siRNA treatment on B cell differentiation was reported when compared with the effects of the PTPN22 620W allele in patients. 34 This further supports the gain-of-function hypothesis. As initially suggested by Dai et al. in their report using PTPN22 619W mice, 33 Wang et al. recently proposed that the 620W variant might be an altered-function, rather than a loss- or gain-of function variant, 35 proposing altered substrate specificity of this variant as a possible mechanism for the observed effects. 35 Although convincing, this hypothesis still lacks further evidence to draw a final conclusion.
In addition to the two already mentioned variants, two far less studied polymorphisms in PTPN22 have been reported. One is a rare missense variant that results in a substitution of Histidine 370 with a Glutamine residue (H370N variant) reported by Rivas et al., the other an SNP in the promoter region of PTPN22 (SNP ID rs2488457). The H370N variant might result in increased risk to develop IBD, 36 but apart from the study by Rivas et al., this variant has not been described further and nothing is known about the functional consequences of the H370N variant. Presence of rs2488457 on the other hand has been reported to enhance the risk of developing rheumatoid arthritis in Asian populations,37,38 but seems not to have important effects in Caucasians. 39 Also for rs2488457 very little is known about functional consequences. We will not address these two less described variants in this review.
The role of PTPN22 in lymphocytes
PTPN22 is expressed in both myeloid and lymphoid immune cells, but barely present in non-hematopoietic cell types. 40 Given the strong association with inflammatory diseases, several studies addressed the role of PTPN22 in immune cell signalling. In T cells, PTPN22 activity attenuates T cell antigen receptor (TCR) signalling by interacting with and de-phosphorylation of the TCR associated kinases lymphocyte-specific protein tyrosine kinase (Lck) and zeta-chain associated protein (ZAP)-70.41,42 Further interaction partner of PTPN22 in the TCR signalling cascade include VAV, and Csk.42,43 As described above, interaction with Csk is suggested to have important consequences on the ability of PTPN22 to suppress TCR signalling. 26 While Csk sequesters wild-type PTPN22 and prevents binding of PTPN22 to its substrates, Csk cannot bind PTPN22 620W, which finally results in increased suppression of TCR signalling. 26 Consistent with the gain-of-function theory, presence of the 620W variant results in reduced T cell receptor signalling in T1D patients and healthy individuals, 29 while PTPN22 deficient T cells are hyper-responsive to TCR ligation. 44 Further, overexpression of PTPN22 in the T cell compartment results in attenuated TCR signalling, and in a non-obese diabetic (NOD) mouse model is able to reduce autoimmune diabetes incidence. 45 PTPN22 deficient mice in contrast display increased levels of effector and memory T cells, but interestingly do not show signs of autoimmunity. 44 Of note, the same phenotype on effector/memory T cells has been observed in mice expressing the 619W variant in PTPN22, an aspect arguing for a loss-of-function effect of the variant.32,33 The lack of autoimmunity in PTPN22 deficient mice was explained in additional studies, which revealed that these mice harbour increased numbers of regulatory T cells.46,47 The study by Brownlie et al. reported that PTPN22 deficient Treg have enhanced suppressor functions and thereby are able to counteract the increased T cell activation found in PTPN22 deficient effector T cells. 46 However, the study by Maine et al. did not find intrinsic differences in Treg function. 47 While Maine et al., attributed the increased number of Treg to an enhanced thymic output of Treg, 47 Brownlie et al. did not see differences in Treg output from the thymus, but rather suggested increased proliferation/induction of peripheral Treg as a source for enhanced Treg numbers. 46 Of note, mice expressing the PTPN22 619W variant showed no difference in Treg induction or regulatory capacity, 33 an effect arguing against the loss-of-function theory.
In a further study to investigate the role of PTPN22 in autoimmunity, Maine et al. addressed the effect of PTPN22 deficiency on germinal centre reactions, where B cells are activated by follicular T helper cells (Tfh) to differentiate into antibody producing plasma cells. In this study, they found increased numbers of Tfh cells, but in contrast to conventional Treg, follicular regulatory cells (Tfr) were not enhanced. Ultimately, this resulted in increased B cell activation and antibody production, mediating pronounced arthritis severity in a mouse model engineered to develop T helper cell dependent arthritis. 48 However, in a mouse model of SLE, PTPN22 deficiency had no effect on disease incidence or severity, although PTPN22 deficient animals still showed enhanced levels of Tfh cells and enhanced antibody production. 49 In general, the effect of PTPN22 on autoimmunity seems to depend on the genetic background, as presence of PTPN22 619W results in signs of systemic autoimmunity in a mixed genetic background, but is lost when the mice are backcrossed into a pure C57BL/6 background. 33
Presence of the PTPN22 620W variant leads to an increased frequency of auto-reactive B cell clones in healthy patients50,51 and T1D patients. 51 Several studies argue that this is the result of B cell intrinsic defects, 21 such as enhanced survival of auto-reactive B cells at the transition from immature to naive B cells50,51 due to alterations in B cell receptor (BCR) signalling.50,51 A more recent study however claims that this effect might result from changes in follicular Th cell activation, which drives altered activation of B cells in germinal centres, rather than from a B cell intrinsic effect. 48 Nevertheless, Dai et al. demonstrated that B cell specific overexpression of the PTPN22 619W variant suffices to induce enhanced GC proliferation and the emergence of autoimmunity in a mixed genetic background, 33 clearly indicating a B cell intrinsic role for PTPN22. In contrast, the study by Maine et al. nicely shows that follicular T helper cells are important drivers of aberrant germinal centre B cell development in PTPN22 deficient mice. 48
Recently, an important role for innate type lymphoid cells (ILC) has been implicated in the pathogenesis of intestinal inflammation. 52 So far, very little is known about the role of PTPN22 in these cells. Although very high expression of PTPN22 is found in NK cells, 40 which are regarded as one subset of ILCs, 53 the consequences of altered PTPN22 function in NK cells, or other types of ILCs remains elusive. It is also not known if other ILCs beside NK cells express PTPN22.
PTPN22 in myeloid immune cells
Most studies addressing PTPN22 function have been carried out in lymphocytes, but PTPN22 expression is very high in other immune cell types as well, including neutrophils, macrophages, and dendritic cells (DC).21,40 We have demonstrated that loss of PTPN22 in monocytes results in profound changes in IFN-γ-induced signalling with reduced STAT1, but strongly enhanced p38 activation, ultimately resulting in drastically enhanced IL-6 secretion together with reduced IL-12 levels.
54
Loss of PTPN22 resulted in an altered response to the bacterial product muramyl-dipetide (MDP) causing not only enhanced autophagy induction but also increased secretion of pro-inflammatory IL-6 and IL-8.
55
Hence loss of PTPN22 in myeloid immune cells results in an increase in the secretion of pro-inflammatory cytokines (Figure 1).
PTPN22 controls IFN-γ and MDP induced signalling cascades.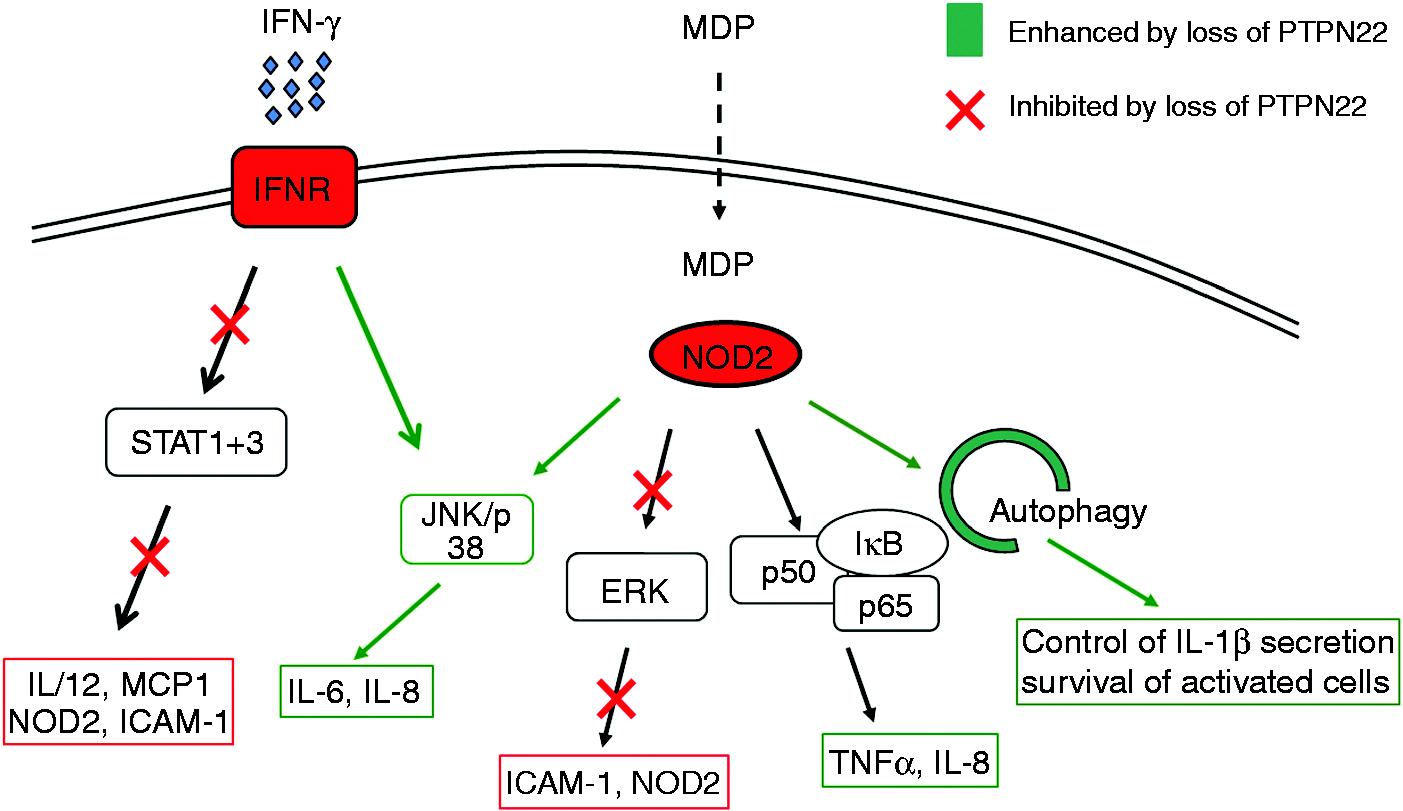
Wang et al. demonstrated that presence of the PTPN22 620W variant results in impaired type-1 IFN production upon TLR4 ligation. 35 Of note, this effect seems to be independent of PTPN22 phosphatase activity but relies on direct interaction of PTPN22 with TRAF3, which represents the most important signalling molecule in terms of type-1 IFN induction downstream of TLR4. 56 While the common PTPN22 variant interacts with TRAF3 and mediates its K63 ubiquitination, cells expressing the disease-associated 620W variant fail to induce TRAF3 ubiquitination upon TLR4 ligation, which ultimately results in decreased type-1 IFN production. 35 Consistent with the fact that direct interaction of PTPN22 and TRAF3 is required for efficient type-I IFN production, similar effects were observed in PTPN22 deficient cells. 35 This indicates a loss-of-function effect of the variant in this particular aspect of PTPN22 function. The effect of loss of PTPN22 on type-1 IFN production was confirmed in an additional study, where enhanced IFN-α production was found upon TLR7 ligation in PTPN22 deficient plasmacytoid dendritic cells. 49 While Wang et al. did not find a difference in NF-κB-mediated cytokine secretion upon TLR4 ligation, 35 in our hands, loss of PTPN22 resulted in increased secretion of NF-κB-mediated IL-8 upon MDP treatment. 55 This discrepancy cannot be fully explained with the current body of knowledge, but might result from differences in the activation of additional signalling pathways that indirectly influence NF-κB activity.
Further studies demonstrated that loss of PTPN22, as well as the presence of the 620W variant, promotes differentiation of monocytes into pro-inflammatory macrophages, 57 and DC from PTPN22 mice expressing the 619W variant expressed higher levels of the co-stimulatory molecule CD40 and induced higher proliferation of OT-II T cells in in vitro co-culture experiments. 32
PTPN22 in IBD patients
The PTPN22 620W variant is regarded as a general autoimmune risk allele, as it is associated with increased risk to develop several autoimmune disorders, such as RA, SLE or T1D.
20
Interestingly, presence of the PTPN22 620W variant at the same time results in a reduced risk to develop CD.
15
This opposite effect on intestinal inflammation in contrast to autoimmune diseases evokes the question about the underlying mechanism(s). We found that PTPN22 expression is reduced in the intestine of both UC and CD patients. This reduction was found to be mainly due to a dominant down-regulation of PTPN22 in cells of the monocyte/macrophage linage (CD68+ cells), whereas we only observed a subtle difference in PTPN22 expression in T or B cells.
54
We further showed that TNFα and IL-1β, two cytokines highly elevated in active lesions of IBD patients, mediate reduction of PTPN22 levels in monocytes/macrophages,
54
however, the mechanism of this down-regulation still remains elusive. The specific reduction of PTPN22 in the myeloid cell compartment in the intestine of IBD patients indicates an important role of PTPN22 in these cells for maintaining intestinal homeostasis (Figure 2). On the other hand, in classical autoimmune diseases, such as RA, SLE or T1D, T and B cells play a predominant role.58,59 This contrasting contribution of the innate versus the adaptive arm of the immune system to disease onset might explain why PTPN22 variants reduce the risk for IBD, while in classical autoimmune disorders the same variant is associated with an enhanced risk.
Reduced PTPN22 expression in IBD patients.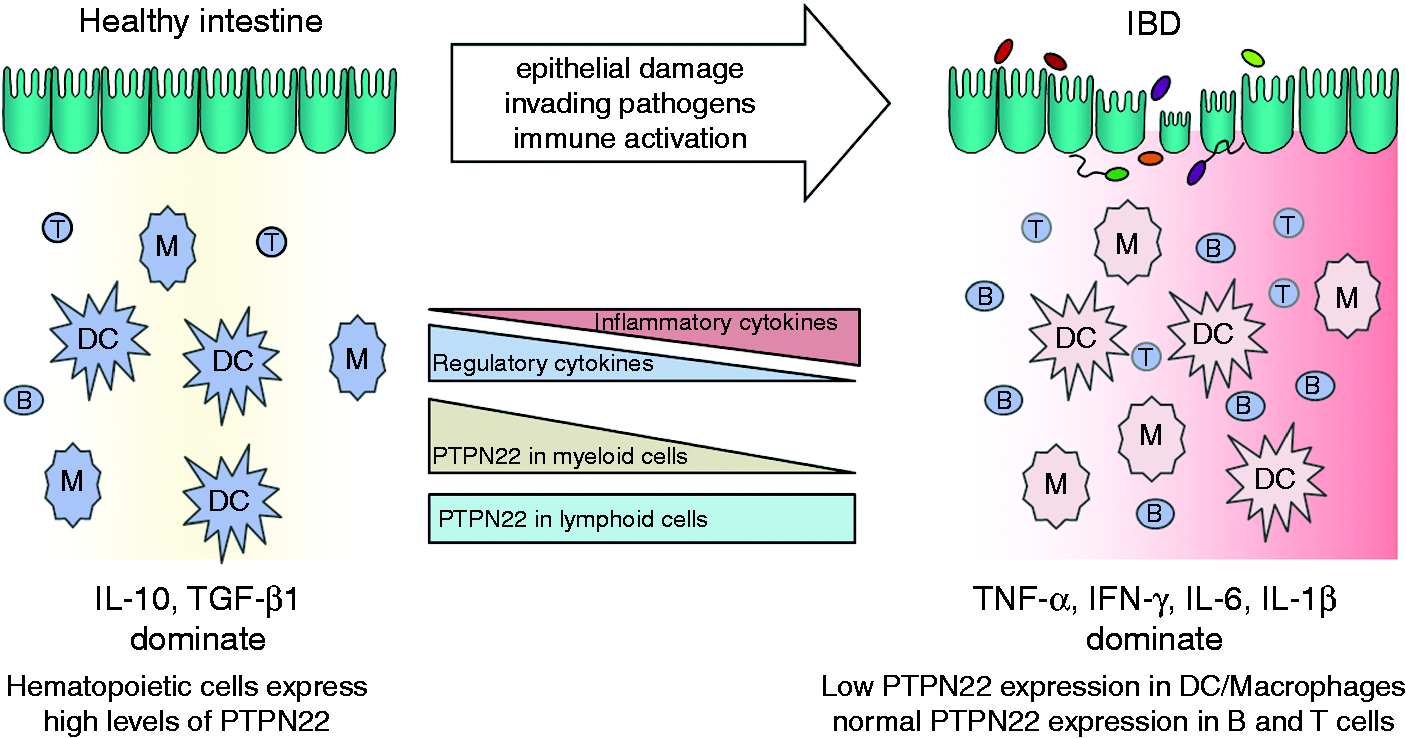
PTPN22 in experimental colitis
Although several genetic association studies revealed an association between PTPN22 and IBD susceptibility,15,60 only a few studies addressed its functional role during intestinal inflammation. One study showed that in a T cell mediated colitis model, where naive T cells were transferred into immune-deficient mice, naive PTPN22 deficient T cells induced aggravated intestinal inflammation and tissue damage. 46 Interestingly, transfer of PTPN22 deficient regulatory T cells – but not transfer of wild-type regulatory T cells – rescued this phenotype. 46 This effect is attributed to increased suppressive competence of PTPN22 deficient regulatory T cells. 46 A recent study further revealed that PTPN22 deficient naive T cells proliferate faster, acquire full effector functions and lose self-tolerance in immune deficient hosts. 61 This explains how PTPN22 deficient T cells are able to mediate increased inflammation when injected into lymphopenic mice.
The important protective role of PTPN22 during intestinal inflammation has further been confirmed in two independent studies using a dextran-sodium sulphate (DSS) induced model of acute colitis.35,57 In both studies, loss of PTPN22 resulted in increased reaction to DSS with aggravated weight loss and enhanced colitis severity.35,57 The enhanced colitis susceptibility has been attributed to altered TLR4 signalling and reduced type I interferon production in one study, 35 or altered macrophage polarization in the other report, respectively. 57 As type I interferon protect mice from colitis, 62 the effect of loss of PTPN22 on type I interferon might – at least in part – explain why PTPN22 deficient mice suffer from more severe colitis and how reduction of PTPN22, as observed in IBD patients, might contribute to disease pathogenesis. However, the cell-type specific contribution to intestinal inflammation has not been directly addressed. We have observed that PTPN22 deficiency results in reduced myelin peroxidase activation and decreased granulocyte infiltration into the inflamed colon during DSS colitis (author’s unpublished data), suggesting a role for PTPN22 in mediating granulocyte function/recruitment.
As PTPN22 expression is reduced mainly in myeloid cells within the intestine of IBD patients, 54 transfer of PTPN22 deficient T cells or colitis induction in a setting where PTPN22 is missing in all cells, might be an incomplete approach to address PTPN22 function during intestinal inflammation. Additional in vivo studies including PTPN22 deficiency specifically in non-lymphoid immune cells would greatly improve our general understanding of the role of PTPN22 in IBD.
In summary, IBD patients feature decreased levels of PTPN22 in myeloid cells in the intestine.
54
Several studies demonstrated that loss of PTPN22 in myeloid cells changes the response towards pro-inflammatory cytokines and bacterial products.35,54,55,57 Hence, reduction of PTPN22, as observed in IBD patients importantly affects the cytokine balance within the inflamed intestine, ultimately promoting disease persistence and progression (Figure 3).
Proposed mechanism showing how PTPN22 is involved in IBD pathogenesis.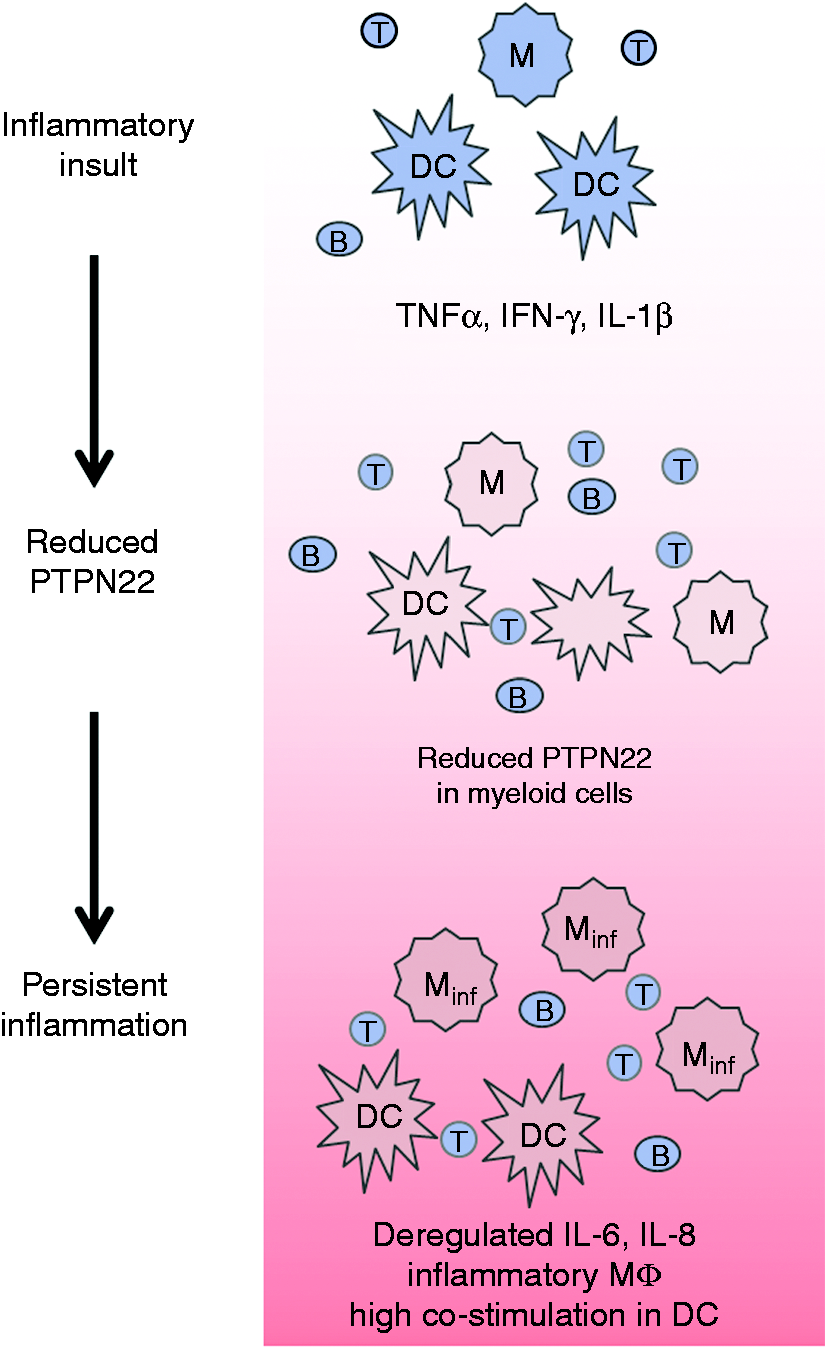
Future directions and concluding remarks
PTPN22 is critically involved in controlling immune cell activation and thereby plays an important role in maintaining intestinal homeostasis. Although in B and T cells the mechanism showing how PTPN22 affects cell signalling pathways is well studied, its role in myeloid cells remains less defined, and very little is known about its function in ILC. Regulation of the innate immune system plays an essential role in the intestine, and levels of PTPN22 in myeloid cells are drastically reduced in the intestine of IBD patients. Therefore, future studies to define the role of PTPN22 in myeloid cells, as well as in innate lymphoid cells will clearly enhance our understanding how PTPN22 contributes to intestinal homeostasis.
Footnotes
Conflict of interest
None declared.
Funding
This work was supported by research grants from the Swiss National Science Foundation (SNF) to MS (grant number 314730-146204 and grant number CRSII3_154488/1).