
Abstract
Two decades ago, the first reports of the use of monoclonal antibodies targeting tumour-necrosis factor α heralded a revolution in treatment options for moderate to severe Crohn’s disease and ulcerative colitis. Nonetheless, patients with refractory disease or loss of treatment response are all too familiar to gastroenterologists. Preventing the infiltration of the gastrointestinal mucosa by circulating cells of the immune system using antibodies targeting the adhesion molecules involved represents an attractive new treatment option. Vedolizumab has recently received European and US regulatory approval for treatment of ulcerative colitis and Crohn’s disease on the basis of encouraging results from one of the largest phase III trial programmes ever conducted in the field of inflammatory bowel diseases and promising safety data. Are we now seeing another revolution in the management of inflammatory bowel disease, and how can this new drug best be used in clinical practice?
Keywords
In May 2014 the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) both granted wide-ranging approval for the marketing of vedolizumab (VDZ) as Entyvio for the treatment of Crohn’s disease (CD) and ulcerative colitis (UC), thus bringing to a close an approvals process lasting 14 years and a scientific development process spanning three decades. VDZ is a humanized version of a murine antibody first described by Dr Andrew Lazarovits in the laboratory of Dr Robert Colvin at the Massachusetts General Hospital, Boston, as reacting with an uncharacterized late-stage marker of lymphocyte activation. 1 With remarkable insight in recognizing a potential therapeutic target, over the subsequent decade dedicated work by Lazarovits determined the antigenic target as the integrin formed through heterodimerization of the α4 and β7 integrin chains (α4β7), characteristically expressed by gut-homing lymphocytes. 2 Development of a humanized form of the antibody proceeded under licence by LeukoSite, a Cambridge, Massachusetts based biotech start-up, and resulted in the demonstration of therapeutic benefit in a non-human primate model of chronic colitis. 3 After the 1999 acquisition of LeukoSite by Millennium Pharmaceuticals, another Cambridge, Massachusetts biotech company, human clinical trials of VDZ commenced in inflammatory bowel disease (IBD) patients. Finally, Takeda’s 2008 acquisition of Millennium brought the drug into the Takeda portfolio, from which it will now be marketed.
The VDZ target: α4β7 integrin
Integrins are cell surface adhesion molecules, formed from the heterodimerization of one α-subunit and one β-subunit, and vital to the complex process of leukocyte extravasation, whereby cells circulating in the vasculature first tether to the vascular endothelium via weak interactions involving selectins. Arrest of a leukocyte and subsequent exit from the circulation and tissue infiltration will depend upon the expression of integrins upon the cell surface capable of binding to endothelial ligands – a process that is dependent upon the activation state of both the leukocyte and the endothelium, both of which may be influenced by local and systemic inflammatory mediators.
B lymphocytes leaving the bone marrow and T lymphocytes exiting the thymus are in a so-called ‘naïve’ state, and recirculate through the peripheral blood lacking the ability to infiltrate tissues. Upon encounter with their cognate antigen in the context of a draining lymph node and under suitable inflammatory conditions, both cell types may undergo clonal expansion. During this process, cell surface selectin and integrin expression changes, permitting the activated lymphocyte to exit from the lymph node and infiltrate those inflamed tissues where the vascular endothelium expresses suitable ligands. Upon termination of the immune response, most of these effector lymphocytes will undergo cell death through apoptosis, but a small population will remain in the circulation or in the tissue as memory cells. This memory population is defined by altered expression of surface markers, including integrins and selectins that define specificity of tissue distribution. Importantly, the context in which the naïve lymphocyte first encounters its antigen will determine the pattern of integrin upregulation and the tissue specificity of the subsequent memory population formed. In murine studies, the upregulation of α4β7 on T cells and acquisition of the capacity to infiltrate the gastrointestinal mucosa occurs uniquely upon those T cells encountering their antigen in the context of retinoic acid producing dendritic cells from the mesenteric lymph nodes, but not when cognate antigen is presented by dendritic cells from other locales.4,5
Integrin α4β7 expression is thus restricted to specific populations of lymphocytes (Figure 1). Strong staining is observed on those lymphocytes resident in tissues throughout the human gastrointestinal tract.
6
Both CD4+ and CD8+ T cells in the lamina propria have been reported to show high levels of α4β7 expression, whilst T cells in the intraepithelial layer show minimal α4β7 expression.7,8 Multiple other immune cell populations are present within the gastrointestinal mucosa that play roles in IBD pathogenesis, including macrophages, natural killer cells and innate lymphoid cells.
9
The recirculation of these cells in humans, their expression of α4β7, and hence the potential for VDZ to disrupt their biology, all remain poorly understood and require further characterization.
Distribution of α4β7 integrin. The vedolizumab (VDZ) target integrin α4β7 is expressed on a range of cell types. These include cells resident in the tissues (particularly T cells of the intestinal lamina propria, with scant data for other cell types). In the peripheral circulation, the highest reported expression is on a subset of CD4+ memory T cells (CD45RO+ integrin α4β1−), but a range of other cell types also express α4β7 on at least a subset of their total number. The expression of α4β7 on precursor leukocyte cell populations has been reported in murine embryonic and adult tissue, but has been little studied in humans. Binding of VDZ to α4β7 integrin will disrupt the interaction with mucosal vascular addressin cell adhesion molecule 1 (MAdCAM-1), an adhesion molecule expressed on the surface of the vascular endothelium in small intestinal and colonic lamina propria venules, and in Peyer’s patches. VDZ may also disrupt fibronectin binding in the extracellular matrix, and also induce so-called ‘outside-in’ signalling through the α4β7 integrin, though this hypothesis remains to be formally tested.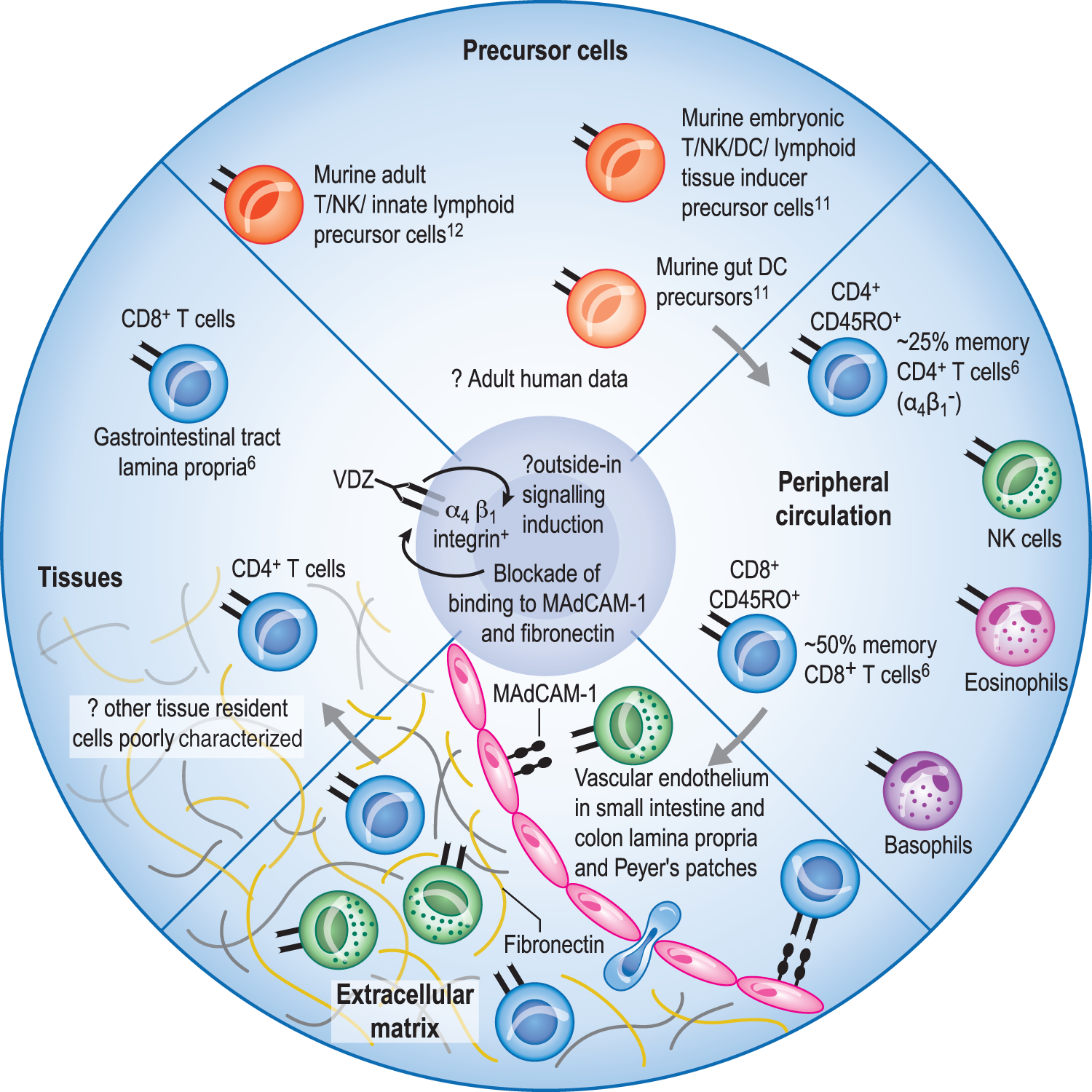
In the peripheral circulation, the highest expression level of α4β7 is observed on a subpopulation of approximately 25% of the memory subset of CD4+ T cells. 6 Less intense expression is also observed on a subset of memory CD8+ T cells, as well as most peripheral blood B cells. As expected, naïve CD4+ and CD8+ T cells show very low surface expression of α4β7. A range of other cell types, including natural killer (NK) cells, basophils and eosinophils, express varying levels of α4β7 on the surface of a small subset, whilst monocytes and neutrophils are negative for surface expression. Interestingly, α4β7 is expressed on murine eosinophils, and β7-integrin knock-out mice show no difference in pulmonary eosinophil recruitment in airway challenge models, but decreased eosinophil recruitment to gastrointestinal inflammatory stimuli. 10
α4β7 may also be transiently expressed during the development of immunocyte precursors. Expression of α4β7 defines a pluripotent population of cells during murine embryogenesis with the capacity to differentiate into T, NK or dendritic cells, or to induce formation of intestinal Peyer’s patches. 11 In adult mice, bone marrow cells expressing α4β7 can give rise to T cells, NK cells and innate lymphoid cells. 12 Likewise, a murine α4β7+ cell population gives rise to dendritic cells resident in the lamina propria. 13 In the absence of this cell population in a T cell transfer model of colitis, T cells homing to the gut fail to develop appropriate regulatory subsets, leading to increased inflammation. 14 Whilst data on human precursor populations are more limited, the widespread expression of α4β7 mandates future consideration of the effects (and any adverse effects) of VDZ from a broad perspective beyond simply that of memory T cell blockade. 15
Integrin–ligand binding and VDZ
A key ligand for integrin α4β7 is mucosal vascular addressin cell adhesion molecule 1 (MAdCAM-1), which is expressed on the endothelium of venules in the lamina propria of the small intestine and colon, as well as in Peyer’s patches. 16 Endothelial MAdCAM-1 expression is upregulated by a variety of stimuli, including tumour-necrosis factor (TNFα), and colonic tissue from patients with active UC and CD shows increased MAdCAM-1 immunostaining.16,17 Other reported target ligands for the α4β7 integrin include the major extracellular glycoprotein fibronectin. In vitro, VDZ disrupts the binding of α4β7+ cells to fibronectin at a similar sub-nanomolar concentration to the disruption of binding to MAdCAM-1. 6 The significance of this in vivo is unclear, due to the existence of multiple other fibronectin binding partners, and thus the primary mechanism of action of VDZ likely remains disruption of α4β7/MAdCAM-1 interactions. By binding with high affinity to α4β7, the only known ligand for MAdCAM-1, VDZ disrupts the α4β7/MAdCAM-1 interaction, hence blocking a key step in the infiltration of α4β7+ cells into the gut.
Integrin functions extend beyond mechanical coupling of cells to endothelium. Motifs in the carboxy-terminal tails of the α and β integrin subunits may engage intracellular signalling cascades with subsequent modulation of cellular activation (so-called ‘outside-in’ integrin signalling). 18 Likewise, changes in cellular activation state and surrounding chemokine milieu are transduced through signalling pathways, leading to modifications to key proteins binding integrin intracellular domains, leading to conformational changes in the extracellular domains from low-affinity to high-affinity ligand binding states (so-called ‘inside-out’ integrin signalling). 18 The extent and significance of VDZ disruption of these bidirectional signalling pathways in α4β7+ cells remain to be characterized, but may underlie some of the clinical effects observed.
Understanding the relative importance of the mechanisms of action of VDZ, including cellular targets, disruption of MAdCAM-1 binding, fibronectin binding and modulation of integrin-mediated signalling, is of more than academic importance, since in the coming years VDZ will likely be joined by a range of other anti-integrin therapeutics currently at varying stages of development for IBD, all differing in their molecular target. These include etrolizumab, a monoclonal antibody targeting the β7 integrin subunit with activity against both the α4β7 integrin as well as the αEβ7 integrin, important for retention of T cells within the mucosal intraepithelial compartment; 19 AMG 181, an anti-α4β7 monoclonal delivered subcutaneously; PF-00547659, a monoclonal antibody against MAdCAM-1; and AJM300, an oral α4 integrin small molecule antagonist. A current phase I trial is also evaluating the pharmacokinetics of VDZ given subcutaneously, offering the promise of a future alternative to intravenous VDZ administration. 20
VDZ and IBD
The clinical effects of VDZ in IBD have been demonstrated in a series of clinical studies, culminating in the phase III GEMINI studies.21–23 These placebo-controlled studies were designed first to assess the efficacy and safety of VDZ in the induction of clinical response and remission in individuals with moderate to severe active UC and CD, and then to assess the efficacy and safety of VDZ in the maintenance of remission in those individuals achieving an initial response to therapy.
Studies in both patient populations followed a similar design (Figure 2). An initial cohort in each study was randomized to VDZ 300mg given as an intravenous dose at week 0 and week 2, with the primary clinical endpoints evaluated at week 6. Those patients exposed to VDZ and meeting response criteria at week 6 were then re-randomized into maintenance studies, receiving either placebo every four weeks (Q4W), VDZ infusions Q4W, or VDZ infusions every eight weeks (Q8W, with additional placebo infusions to maintain blinding). Those patients who received placebo induction therapy continued to receive placebo during the maintenance studies. In order to fulfil sample-size criteria for the maintenance studies, an additional cohort of individuals in each study was given open-label VDZ induction therapy, with those who met week 6 clinical response criteria then undergoing re-randomization to VDZ or placebo for the maintenance phase. Those patients in either cohort who received VDZ induction therapy but failed to meet week 6 response criteria were not re-randomized, but continued to receive VDZ Q4W. These patients were not included in the intention-to-treat analyses, but were included in safety data and in exploratory clinical outcome analyses. Both UC and CD maintenance studies reported outcomes at 52 weeks, with those patients deemed to have derived clinical benefit then invited to enter an ongoing, open-label, long-term safety and efficacy follow-up study (GEMINI-LTS).
Design of GEMINI studies. For the induction studies in GEMINI 1, 2 and 3, patients were randomized to vedolizumab (VDZ) dosing at week 0 and week 2, or placebo (PBO). Outcome analysis was performed on an intention-to-treat basis at week 6. GEMINI 1 and 2 also included maintenance studies. For these studies patients responding to VDZ at week 6 were re-randomized to VDZ every four weeks (Q4W), PBO Q4W, or VDZ every eight weeks (Q8W) (with alternate PBO infusions to maintain full blinding). In order to achieve sufficient statistical power, this re-randomized responder group comprised responders from the induction studies (cohort 1) as well as responders from an open-label group given VDZ at week 0 and week 2 (cohort 2). Primary and secondary outcomes were assessed on an intention-to-treat basis in these patients at week 52. Non-responders from both cohorts could continue to receive VDZ Q4W, whilst PBO patients from the induction study continued to receive PBO during the maintenance study. Neither of these two groups was included in the main intention-to-treat analysis, but both contributed data to the safety analyses and exploratory clinical analyses.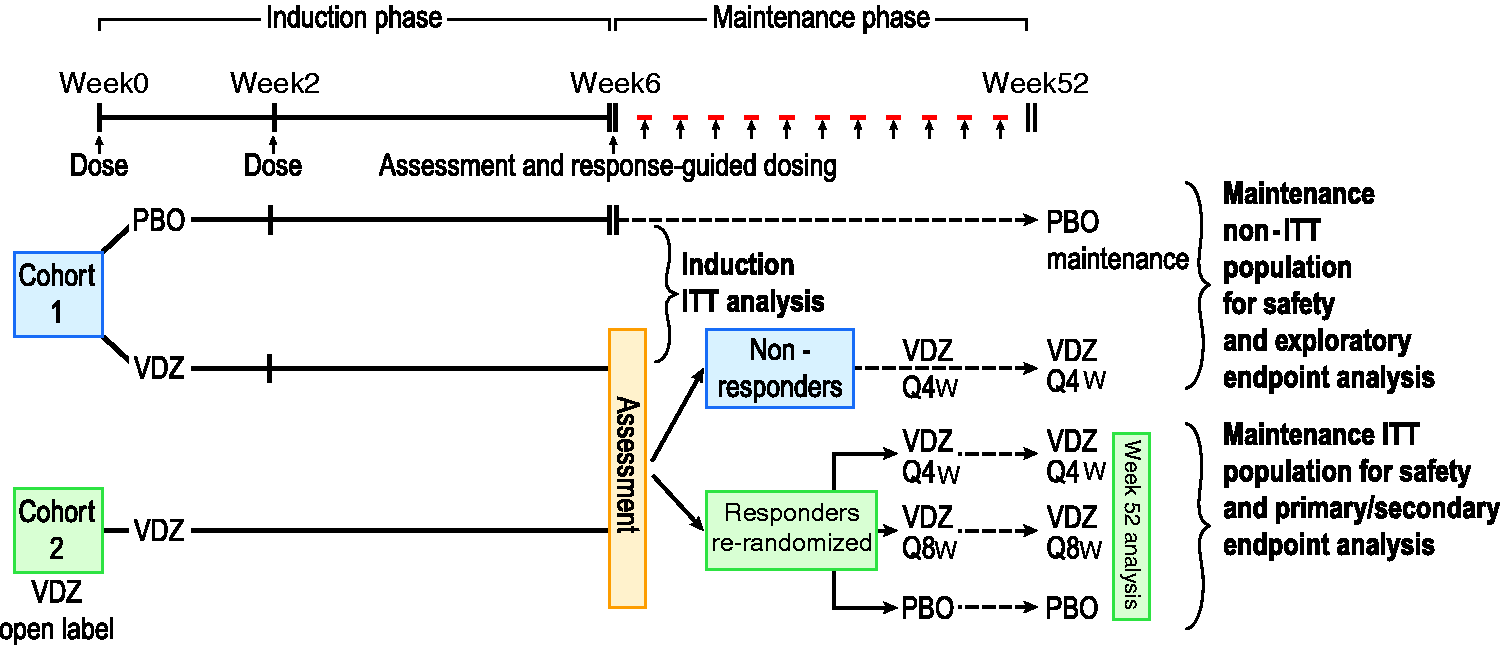
All subjects had previously failed conventional therapies for their IBD. Due to FDA concerns over the risk of progressive multifocal leukoencephalopathy (PML; see below), US subjects were required to have demonstrated an inadequate response, intolerance or loss of response to immunomodulator therapy or to a TNFα antagonist. Outside the USA, corticosteroid dependence, corticosteroid intolerance or an inadequate response to corticosteroids were alternative inclusion criteria, thus the final studies included 17–18% of patients who had failed or were dependent upon corticosteroids alone. Along similar lines, although concomitant treatment with stable doses of immunomodulators was permitted at induction, patients in the USA were required to discontinue immunomodulators after the induction period, whereas non-USA patients were permitted to continue immunomodulators during maintenance therapy. All patients were permitted to continue stable doses of oral aminosalicylates throughout both induction and maintenance periods. Although stable doses of oral corticosteroids were allowed in all patients at induction, all patients entering the maintenance study were required to initiate steroid weaning according to a pre-specified regimen.
Key outcome data from the GEMINI studies. Data shown are percentages of patients achieving the specified outcome (except for week 6 CRP values in mg/l) based upon intention-to-treat analysis, with corresponding p values based upon Cochran–Mantel–Haenszel chi-squared testing as reported in the index studies. Note that maintenance outcomes are reported only for the cohort of patients achieving a clinical response at week 6, then re-randomized to maintenance studies
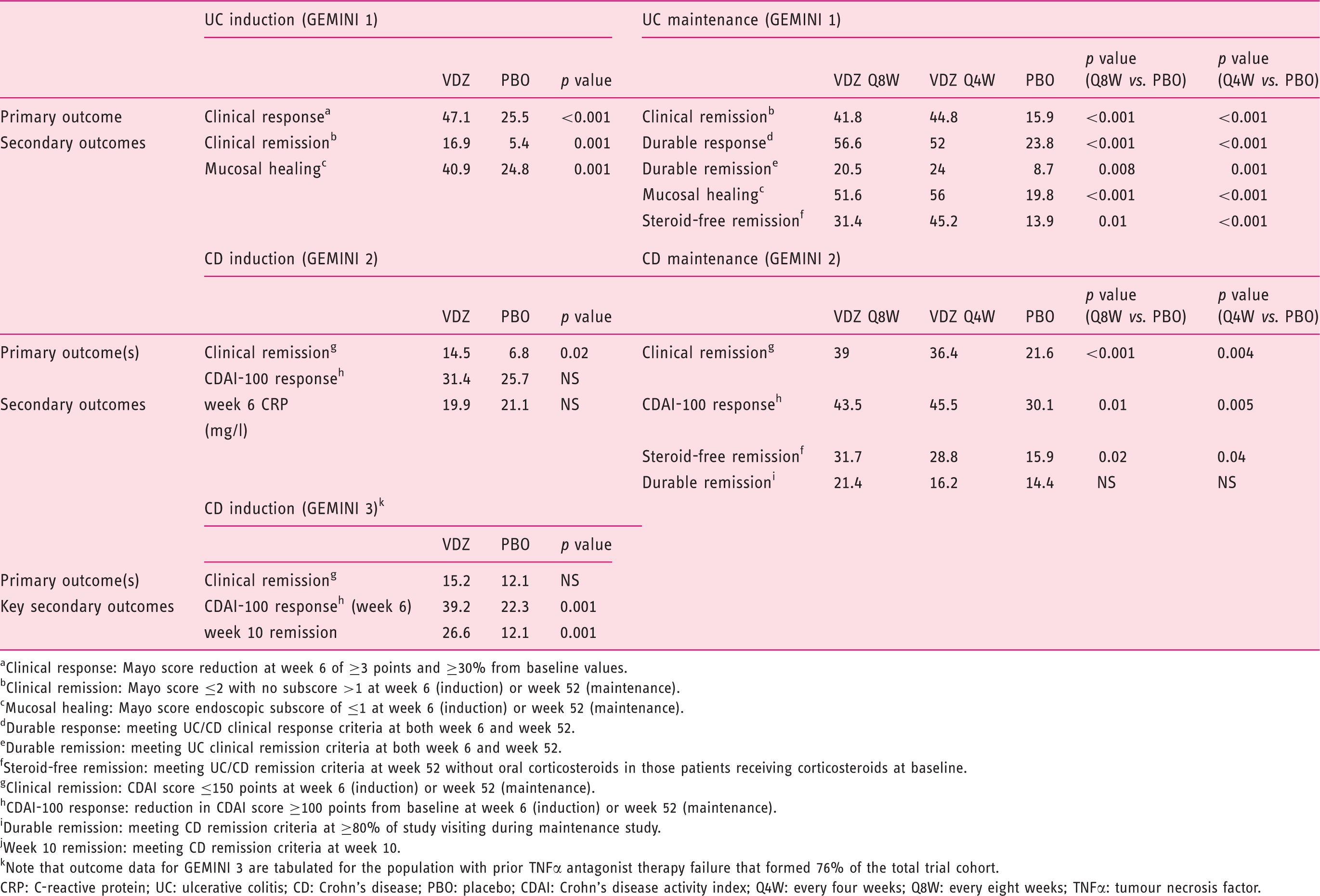
Clinical response: Mayo score reduction at week 6 of ≥3 points and ≥30% from baseline values.
Clinical remission: Mayo score ≤2 with no subscore >1 at week 6 (induction) or week 52 (maintenance).
Mucosal healing: Mayo score endoscopic subscore of ≤1 at week 6 (induction) or week 52 (maintenance).
Durable response: meeting UC/CD clinical response criteria at both week 6 and week 52.
Durable remission: meeting UC clinical remission criteria at both week 6 and week 52.
Steroid-free remission: meeting UC/CD remission criteria at week 52 without oral corticosteroids in those patients receiving corticosteroids at baseline.
Clinical remission: CDAI score ≤150 points at week 6 (induction) or week 52 (maintenance).
CDAI-100 response: reduction in CDAI score ≥100 points from baseline at week 6 (induction) or week 52 (maintenance).
Durable remission: meeting CD remission criteria at ≥80% of study visiting during maintenance study.
Week 10 remission: meeting CD remission criteria at week 10.
Note that outcome data for GEMINI 3 are tabulated for the population with prior TNFα antagonist therapy failure that formed 76% of the total trial cohort.
CRP: C-reactive protein; UC: ulcerative colitis; CD: Crohn’s disease; PBO: placebo; CDAI: Crohn’s disease activity index; Q4W: every four weeks; Q8W: every eight weeks; TNFα: tumour necrosis factor.
Any difference in efficacy of VDZ in UC and CD may reflect differences in immunopathogenesis, and in particular the relative roles of innate immune defects. 9 Indeed, the results bear comparison with the effects of the anti-T cell calcineurin-inhibitor ciclosporin, which demonstrates response rates of up to 80% in severe UC, but has little if any role in the management of CD.24–26 In the same light, the limited data reported for VDZ in fistulizing perianal disease (see Appendices 1 and 2) contrast with the effectiveness of TNFα antagonist therapy;27,28 again, these findings may reflect the importance of non-α4β7+ cells and, in particular, innate immune dysfunction in disease pathogenesis, as well as differences in adhesion molecule expression in perianal mucosa and skin.
There were important differences in the use of endoscopic assessment to define clinical outcomes of the UC and CD studies, with consequences for FDA marketing approval that may be relevant for future study design. By using a complete Mayo score as part of the primary outcome measure (which incorporates endoscopic disease assessment), GEMINI 1 was able to demonstrate improvement of the endoscopic appearance of the mucosa early in UC treatment. This fell short of providing histologic evidence of improvement, and consequently led to insistence by the FDA on the UC licence referring to ‘improving endoscopic appearance’ rather than to ‘mucosal healing’ as originally proposed by Takeda. 29 There is increasing recognition of the importance of endoscopic appearances in predicting disease course, 30 even if cause–effect relationships have not been resolved. In the GEMINI CD studies, no endoscopic outcomes were collected, and no assessment of effects on mucosal appearance can be made.
Since complete Mayo scores were used for the outcome measures of GEMINI 1, and due to the impracticality of performing more frequent endoscopies, GEMINI 1 maintenance outcomes were restricted to disease evaluation at a single week 52 timepoint, with only partial data reporting during the rest of the period. For GEMINI 2, outcome measures were based on Crohn’s disease activity index (CDAI) scores, which do not require endoscopy and were recorded four-weekly. Thus the maintenance outcome measures for CD incorporated ‘durable’ remission, taking account of serial CDAI scores recorded four-weekly during the study period, in which regard VDZ failed to show statistical benefit over placebo. Arguably, the use of frequent assessments throughout a treatment period offers a more complete and patient-centred view of drug utility as a maintenance therapy. Indeed, in responses to the FDA, Takeda argued that if analysis of the CD remission data was limited to just two timepoints (week 6 and week 52), as was the case for the ‘durable remission’ definition in GEMINI 1, then VDZ appeared superior to placebo. 29 Nonetheless, the FDA took the view that overall VDZ for CD had fallen short of demonstrating ‘maintenance of remission’ and granted approval under the terminology ‘achieving clinical remission’. The difference in the UC and CD FDA decisions, although subtle and undoubtedly fair for VDZ, is sure to be studied by trial designers and highlights important questions around the best use of partial interim disease assessment scores in maintenance drug trials alongside only intermittent endoscopic assessment.
Although the GEMINI trial designs assessed induction therapy response at week 6, Takeda successfully argued during regulatory approval for induction dosing at weeks 0, 2 and 6, with maintenance dosing commenced from week 14 onwards in those deemed to have derived benefit upon clinical assessment at this slightly later timepoint. Due to the potential slower response in CD seen in GEMINI 2 and GEMINI 3 (see Appendix 1), the EMA (but not the FDA) accepted that a further induction dose at week 10 may be used for CD patients who have not shown a beneficial response at this timepoint. Since Q4W and Q8W maintenance strategies in both UC and CD demonstrated similar clinical outcomes and similar saturation of α4β7 integrin binding, the maintenance therapy license is for Q8W dosing. Again, the EMA, but not the FDA has permitted prescribing information to indicate that dose intensification to Q4W infusions may be useful for patients who have lost response to Q8W dosing. In the light of pharmacokinetic data from GEMINI 1 and 2 suggesting maximal receptor occupancy on peripheral blood T cells with Q8W dosing, the rationale for this dose escalation needs consideration. Loss of response to TNFα antagonist therapies is associated with the development of anti-drug antibodies and low trough serum drug levels,31,32 and trough drug level monitoring and anti-drug antibody monitoring alongside intensification of dosing frequency appears useful in maximizing benefit from TNFα antagonist therapies. 33 In this regard, it is interesting that maintenance outcomes in GEMINI 1 and 2 did not show correlation with trough drug levels.21,22,34 Furthermore, in patients completing maintenance therapy with VDZ, transient anti-drug antibodies were reported in 4% of subjects21,22 with persistent positivity seen in just 0.6% of subjects (all of whom failed to achieve remission), suggesting that loss of response secondary to the development of anti-drug antibodies may be a rare event. However, the assay used by the manufacturer to report these values lacks specificity in the face of detectable serum drug concentrations, almost certainly leading to underestimation of the true rate of anti-drug antibodies. 35 Therefore, the best strategy for maintenance dosing and monitoring with VDZ remains an open question, but one that needs addressing with outcome studies using therapeutic drug monitoring and an effective anti-drug antibody assay. Although post-marketing investigator-initiated studies may address this, it is worth noting that the FDA has mandated Takeda to reanalyse banked serum samples from GEMINI 1 and GEMINI 2 using an improved anti-drug antibody assay and report on immunogenicity by March 2017. 35
One interesting set of data from the GEMINI studies that has not been made public is the exploratory efficacy outcomes in the treatment groups who failed to respond to VDZ induction therapy, but who remained on VDZ Q4W during the maintenance phase. The publication of these data would help determine whether there is any possible benefit in continuing further VDZ infusions in patients who fail to demonstrate early evidence of clinical benefit from VDZ. Other clinical considerations around the use of VDZ are discussed in Appendix 2.
Safety of VDZ
Overall safety data from the GEMINI studies were remarkable and encouraging for the low incidence of adverse events in the large population of patients exposed to VDZ. The relatively restricted tissue distribution of MAdCAM-1 should limit the effects of VDZ on trafficking of leukocytes into other sites. This is critical since the development, licensing and now clinical adoption of VDZ have all taken place in the dark shadow cast by natalizumab, a monoclonal antibody targeting the α4 integrin subunit. This subunit is a component not just of the α4β7 integrin, but also of the α4β1 integrin. Vascular cell adhesion molecule 1 (VCAM-1), the ligand for α4β1, is widely expressed and is critical for lymphocyte trafficking into the central nervous system (CNS).36–38 Therefore natalizumab was developed as a treatment both for CD and for multiple sclerosis (MS), with FDA approval for MS granted in 2004 and CD in 2008. As early as 2005, reports emerged of cases of PML in two individuals treated with natalizumab, a number growing to 430 natalizumab recipients by January 2014. 39 PML is a devastating demyelinating disease caused by uncontrolled replication of the JC polyomavirus within the CNS. Whilst JC virus is endemic, affecting up to 90% of healthy individuals, PML is rare and limited to immunosuppressed individuals, including those with acquired immunodeficiency syndrome (AIDS) or haematological malignancies, and transplant recipients. Epidemiological confirmation of association of PML with natalizumab treatment has highlighted the particular increased risk in those patients receiving combination immunosuppression. 40
Since VDZ is selective for the α4β7 integrin heterodimer and does not bind the α4β1 heterodimer, the selective nature of blockade achieved by VDZ, compared with natalizumab, should mitigate against the risk of PML with VDZ treatment – a point the academic authors of the GEMINI trials have stressed repeatedly both to licensing agencies and in the scientific literature.41,42 Certainly, no cases of PML have been observed in over 3000 patients so far treated with VDZ, and careful studies of cerebrospinal fluid of VDZ recipients show no effects on lymphocyte composition. 43 Nonetheless, most of these patients have received relatively short exposure to VDZ, whilst risk of PML with natalizumab increases significantly with exposure beyond two years. 40 Furthermore, the theoretical increased safety with α4β7-restricted integrin blockade relies upon the assumption that PML in natalizumab recipients occurs as a result of loss of CNS control of JC virus reactivation through the α4β1-mediated blockade of lymphocyte infiltration.44,45 This model of PML pathogenesis is credible, but unproven, 46 with JC viral latency established in a restricted range of cell types, including kidney epithelium, bone marrow derived cells 47 and the gastrointestinal epithelium, 48 leading to alternative hypotheses of viral reactivation outside of the CNS with subsequent viral dissemination to the CNS contributing to immunosuppressant related PML. 46 For now, both the FDA and EMA have mandated enhanced monitoring schemes for PML in post-marketing studies as well as clear indication of the PML risks in prescribing information.
If much has been made of the theoretical risk of PML with anti-integrin therapies, less attention has perhaps been given to the development of acute hepatitis observed in four patients given VDZ during the GEMINI programme. All four cases ceased VDZ, received intravenous corticosteroids and recovered. Although causation could not clearly be established in these cases, similar reports of severe drug-induced liver injury have been reported for natalizumab.49,50 Integrin α4β7 plays an important role in trafficking of T cells from the gut to the liver in the portal circulation, particularly under conditions of inflammation, 51 and targeting of this axis may disrupt the balance of T cell subsets, including regulatory T cells infiltrating the liver, resulting in a possible biological mechanism for the observed cases. The FDA has mandated that risks of liver injury are explicitly discussed in US prescribing information and package inserts for VDZ, but the EMA did not feel such measures were required for European markets.
In common with any immunomodulatory strategy, effects on infectious complications are of particular interest. The tissue specificity of α4β7/MAdCAM interactions should mitigate against significant systemic immunosuppression, and indeed the safety data from the GEMINI studies are enormously encouraging, showing only a slight excess of infections, mostly driven by a slight excess of non-serious upper-respiratory tract infections in VDZ recipients.21,22 In this regard, it is interesting to note the outcome of a well designed phase I vaccine challenge study, in which healthy individuals injected with VDZ or placebo received oral cholera vaccine and intramuscular hepatitis B vaccine. Although seroconversion and cholera toxin antibody titres were markedly lower in the VDZ-exposed group, antibody titres to intramuscular challenge were unaffected by VDZ exposure. 52
This confirmation of the specificity of the immune attenuation seen with VDZ is remarkable and underlines the positive safety data from the GEMINI studies. Nonetheless, the inhibition of a humoral immune response to oral challenge is in line with gastrointestinal-specific immunosuppression. The incidence of infections with enteric pathogens in all three GEMINI studies was very small, precluding meaningful statistical analysis, but six cases of Clostridium difficile, three cases of Campylobacter infection and one case of Salmonella infection were reported in patients who received VDZ, with no cases in the combined placebo groups. 34 Given the risks associated with gastrointestinal infections in the patient groups in which VDZ will be used, then the gastrointestinal infection safety data will be an important aspect of GEMINI-LTS and post-marketing surveillance, and should prompt vigilance for timely diagnosis and treatment of gastrointestinal pathogens in individuals receiving VDZ.
Reassuringly, the data available so far do not suggest any increased risk of malignancy with VDZ, but again, numbers are small and long-term follow-up lacking. Given the potential for impairment of gastrointestinal immunosurveillance, it is worth noting the four cases of colon cancer and one appendiceal carcinoid tumour reported in VDZ exposed patients during phase III testing and follow-up, giving an incidence rate for colon cancer of 0.66 per 1000 person-years, which is no more than expected in a population with moderate to severe IBD. 34 As with other biologic therapies, long-term follow-up registry data will be vital in establishing an accurate risk profile for VDZ. Clearly, untreated colitis remains the biggest clinical risk factor for development of colorectal cancer in individuals with inflammatory bowel disease. 53
Conclusions
Physicians and patients alike will welcome the addition of a new drug to the therapeutic landscape in IBD and in coming months initial clinical experience and investigator-initiated studies with VDZ will provide useful data and address important questions for clinical practice. Certainly there are many reasons to be optimistic, particularly given the reassuring safety data for VDZ suggesting low risks of toxicities and immunosuppression. For UC, trial data show a clear, early and sustained benefit of VDZ treatment with a large effect size. Data for CD are less dramatic, but still demonstrate a population level effect that will translate into important clinical benefits for some patients. In common with all existing therapies, we still lack adequate markers and mechanistic insights to predict the populations that will derive sustained benefit. In the coming years VDZ will, hopefully, be joined by other new therapeutics, including alternative anti-integrin therapies, as well as Janus kinase (JAK) inhibitors currently in phase III trials. Rather than the traditional model of sequential failure, one might imagine intelligent use and combination of each class of drug in a patient-specific manner, based upon a detailed understanding of mechanism, side effect profiles and predictive biomarkers to enable true maximization of therapeutic benefit. Only with suitable scientific studies, informed by epidemiological and clinical data, will we be able to build the necessary approaches to achieve complete and lasting remission for our patients.
Footnotes
Funding
This work was supported by the Wellcome Trust (WT091993MA postdoctoral clinical fellowship to TR), European Crohn’s and Colitis Organization (ECCO research grant to TR) and the National Institute of Health Research (NIHR) Biomedical Research Centre award to Addenbrooke’s Hospital/University of Cambridge School of Clinical Medicine.
Conflict of interest
TR reports personal fees and non-financial support from GSK, MSD and Abbvie, and non-financial support from Ferring, Abbott, Shire and Dr Falk, outside the submitted work.
Acknowledgments
TR thanks Arthur Kaser and Andrew Metz for helpful discussions and critical reading of the manuscript.