
Abstract
IgG4-related systemic disease is a recently recognized systemic condition characterized by unique pathological features that can affect a variety of organs. It includes a growing number of medical conditions which have the following features in common: diffuse organ swelling or focal mass formation, sclerosing storiforme (whirl-shaped) fibrosis with a lymphoplasmacytic infiltrate rich in IgG4-bearing plasma cells, as well as elevated levels of serum IgG4. It invariably responds to steroid treatment and is mostly diagnosed in elderly men. Well-known syndromes like Mikulicz's disease of the salivary or lacrimal gland, Küttner's tumour of the submandibular gland, Riedel's thyroiditis, or retroperitoneal fibrosis, as well as novel entities such as autoimmune pancreatitis type 1, are now regarded to be manifestations of this systemic disease. This article provides an overview of the epidemiology, concepts of pathogenesis, clinical presentation, proposed diagnostic approaches, treatment options, and differential diagnosis of IgG4-related disease.
Introduction
Previously recognized conditions that are part of IgG4-related disease
Pathogenesis of IgG4-related disease
As IgG4 has lent its name to this group of diseases it is appropriate to focus on pathological aspects of IgG4. IgG4 is a remarkable immunoglobulin in both structure and function. IgG4 accounts for less than 5% of the total IgG and it function differs from that of other IgG subclasses. Amino acid differences in the second constant domain of IgG4 lead to weak or negligible binding of IgG4 to both C1q and Fcc receptors. As a consequence, IgG4 does not activate the classical complement pathway and therefore plays a limited role in activation of the immune system. 6 Another peculiarity of IgG4 molecules is the so-called Fab-arm exchange. Disulphide bonds between the heavy chains of the molecule are weaker than in other IgG subclasses, leading to dissociation and random recombination under certain circumstances. The result is a variety of bivalent or ‘functionally monovalent’ antibodies which are unable to crosslink antigens, thereby losing the ability to form immune complexes 7 . Noteworthy, IgG4 is able to bind the constant domain of other IgGs which allows it to act as an inactive rheumatoid factor and thus might contribute to its anti-inflammatory properties.
IgG4 production can physiologically be induced by sustained or repeated antigen exposure and, similar to IgE production, the production is predominantly induced by type 2 T-helper cells. To our current understanding, a Th2 immune reaction with activation of regulatory T-cells (T-regs) producing transforming growth factor β and interleukin 10 favours IgG4 production. 8 Several autoantibodies have been identified in patients with IgG4-related syndrome. Although none of them seem to be disease specific or diagnostic, they could be related to common target structures in affected organs. Antibodies specific for lactoferrin, carbonic anhydrase II and IV, pancreatic secretory trypsin inhibitor (PSTI or SPINK1), amylase, heat shock protein 10, and plasminogen-binding protein have been detected, predominantly of the IgG type 1. 9 One concept for the eventual breakdown of immune tolerance is molecular mimicry. Pathogens share common epitopes with their host and can therefore initiate a sustained auto-inflammatory stimulus: e.g. carbonic anhydrase II and plasminogen-binding protein of Helicobacter pylori show homologies with human proteins. 9
The presence of T- and B-cells in the lymphplasmacytic infiltrate of affected organs, the presence of autoantibodies and the abundance of IgG4 producing plasma cells suggest an immunologically mediated pathomechanism. Okazaki et al. proposed a two-step process for the development of IgG4-related disease.
9
Briefly, molecular mimicry and genetic predisposition lead to an initial Th1-dominated immune response against self-antigens and the initiation of tissue damage, which is aggravated by a lack of naïve circulating T-regs. The persistent presence of triggering pathogens maintains activation of the immune system which eventually induces a Th2 and peripheral T-reg response, favouring IgG4 production and fibrosis, the latter leading to disease progression (Figure 1).
Pathogenesis of IgG4-related disease.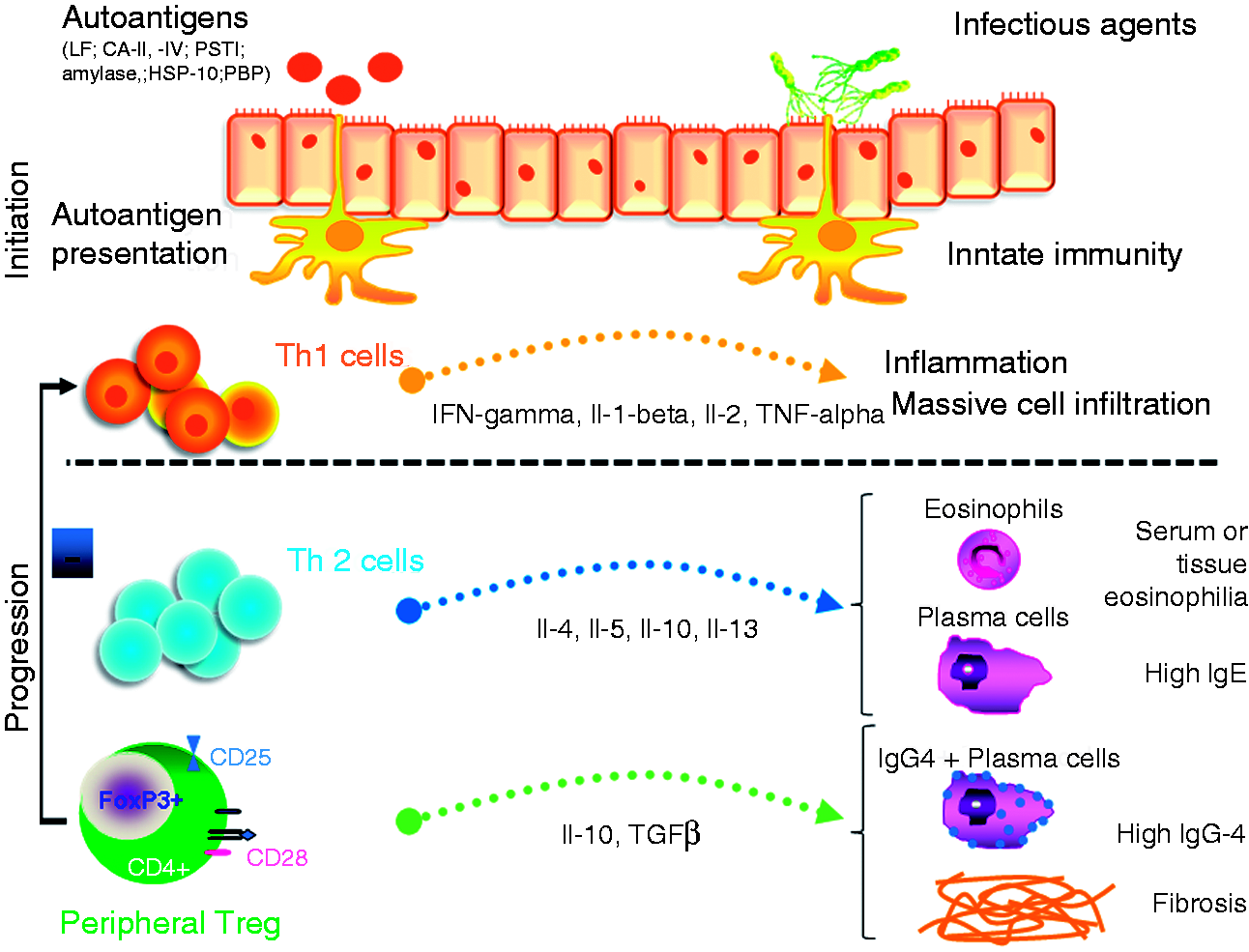
Clinical presentation
With regard to the variety of disease manifestations and for the purpose of this review, we would like to focus on manifestations of IgG4-related disease within the gastrointestinal tract such as AIP type 1 as well as IgG4-associated sclerosing cholangitis. Patients with IgG4-related disease present with diffuse organ swelling or a focal mass formation which leads to either a pseudotumour or nodular organ swelling. The disease can affect multiple organs simultaneously or consecutively, including conditions which have previously been looked at as unique clinical entities (Table 1). The disease usually develops subacutely and most patients feel only moderately ill. Levels of total serum IgG are usually elevated and IgG4 levels more than 25-times above the upper limit of normal have been reported. 10 However, elevation of serum IgG4 over 2- or 3-times the upper limit of normal are generally considered to be specific and diagnostic for AIP.11–17 Other inflammatory serum markers are only moderately elevated and a clinical presentation with weight loss or fever is rather uncommon. 3 About half of the cases present with elevated IgE levels and peripheral blood eosinophilia. 18 Although it has been reported that 74% of cases with AIP type I would undergo spontaneous remission, 19 untreated IgG4-related disease often causes major damage to affected organs, leading eventually to organ dysfunction. 20
The appearance of AIP on imaging has elsewhere been discussed at length.12,14,15 Briefly, the pancreas presents as either generally enlarged giving rise to a so-called ‘sausage-shaped pancreas’ or with a focal mass. These masses often present with a hypodense rim, indicating delayed uptake of contrast enhancers. The pancreatic duct is characterized by calibre irregularities and long stretch narrowing without significant distal dilatation.21–23 The biliary tract is the most frequent site of extrapancreatic involvement of AIP or may occur as a separate entity, then called IgG4-related sclerosing cholangitis. It is best characterized on endoscopic retrograde cholangiopancreatography or magnetic resonance cholangiopancreatography, but also computed tomography or endoscopic ultrasound can reveal typical features such as concentric mural thickening and segmental or long ductal narrowing with and without prestenotic dilatation (Figure 2). Again, the affected soft tissue shows delayed contrast enhancement. IgG4-related cholangitis is subclassified in four different types, depending on the location of the predominant stricture:
24
type 1, located only in the lower part of the common bile duct; type 2, diffusely distributed in the intra- and extrahepatic bile ducts; type 3, stenosis in both the hilar intrahepatic ducts and the lower part of the common bile duct; and type 4, located only in the hilum. Type 2 IgG4-related cholangitis can mimic a group of other pancreaticobiliary disorders on imaging such as pancreatic cancer, primary sclerosing cholangitis, and cholangiocarcinoma
25
and patients frequently present with obstructive jaundice. IgG4-related autoimmune hepatitis can be found in 3.3% of patients previously diagnosed with classical autoimmune hepatitis.26,27 The diagnosis is made in the absence of antimitochondrial, antismooth muscle, and liver kidney microsomal antibodies. Interestingly, 50% of patients with IgG4-related autoimmune hepatitis suffer from acalculous sclerosing cholecystitis. However, therapeutically IgG4-related autoimmune hepatitis is often overlooked because treatment is not different from classical autoimmune hepatitis and symptoms usually respond quickly to steroid treatment.
Typical case of IgG4-related sclerosing cholangitis.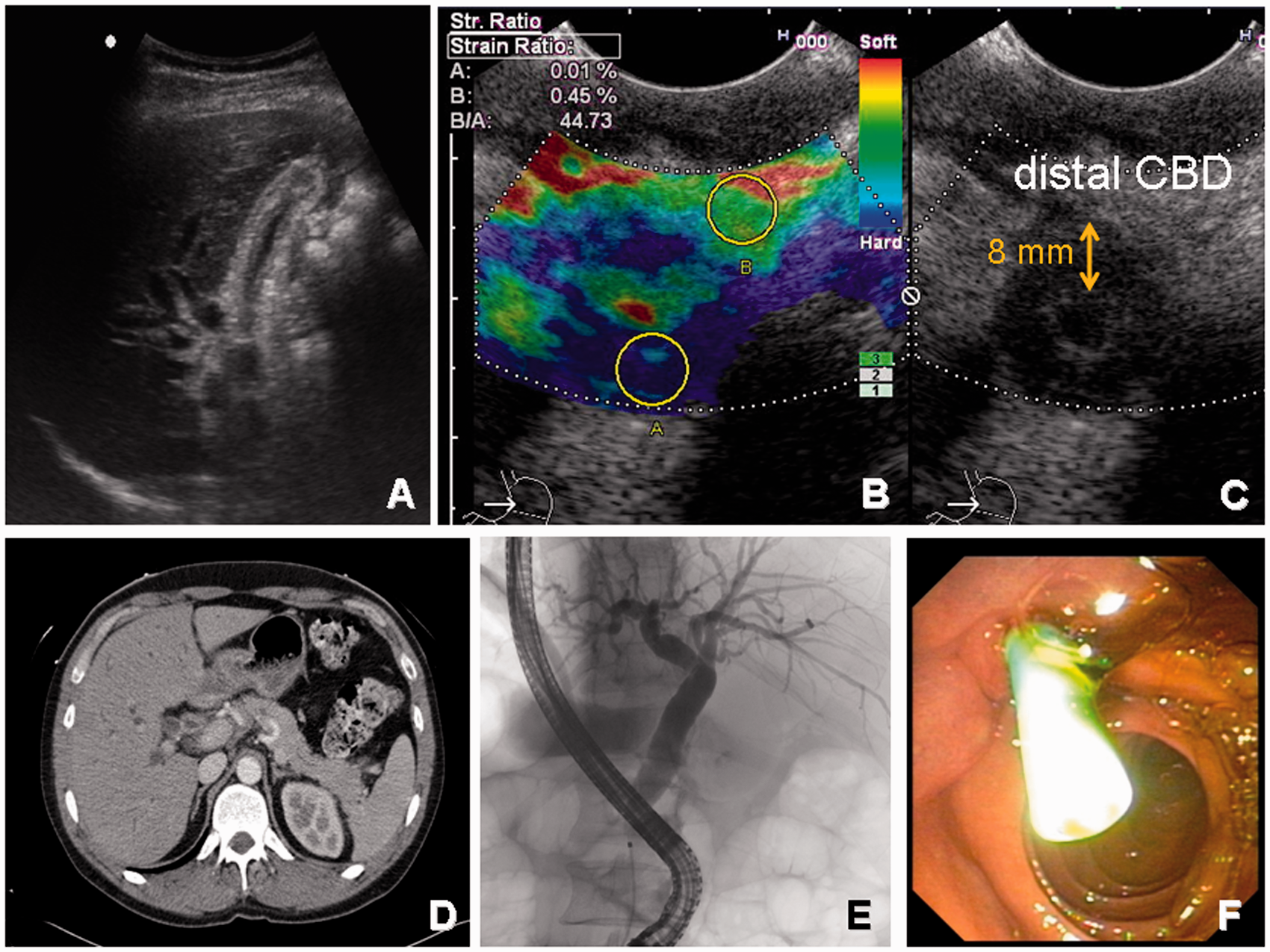
About one-third of patients with IgG4-related syndrome present with tubulo-interstitial nephritis. 7 Lesions are hypoattenuating on computed tomography, show late phase enhancement, and will be iso- or hypointense on T1 and hyperintense on T2-weighted MRI. Cortical, pelvic, and diffuse lesions have been described and are usually present bilaterally and at multiple sites. 25 Similarly, involvement of salivary and lacrimal glands will appear bilaterally as enlarged and homogenous masses. Conditions like chronic sclerosing sialadenitis (Küttner's disease), chronic dacryoadenitis, and Mikulicz's disease as well as Riedel's thyroiditis are now considered manifestations of the IgG4-related syndrome. Retroperitoneal fibrosis (also referred to as morbus Ormond, Ormond's disease) and vascular disease includes similar imaging features, with MRI appearance depending on the current activity of inflammation and oedema. Pulmonary involvement can appear as nodular or focal mass, thickened bronchovascular bundles, air trapping, and mediastinal involvement. Lymphadenopathy is a common feature of IgG4-related disease and can go along with any organ involvement. Nodules are enlarged, with homogenous enhancement and no calcification or necrosis. 25
Histological features
The recognition of distinct histopathological features in a variety of previously independent conditions has finally led to the concept of a systemic IgG4-related disease. The key findings are:
dense lymphoplasmacytic infiltrate with storiform (whirl-shaped) fibrosis obliterative phlebitis abundance of IgG4-bearing plasma cells.
The abundance of IgG4-positive plasma cells is a necessary but not sufficient criterion for the diagnosis of IgG4-related disease. Numbers of more than 10 IgG4-positive cells per high power field and a ratio of IgG4-positive cells to all IgG-bearing cells >40–50% are highly suggestive.5,17,36 Neutrophils are not a common feature and, although the infiltrate tends to aggregate around the ducts of glandular organs, no direct ductal infiltration is seen. B-cells form typical germinal centres, whereas T-cells will be found in a more scattered pattern. Granulomas are uncommon. 7 Histopathological analysis of affected organs remains the gold standard for the diagnosis of IgG4-related disease.
Diagnostic criteria
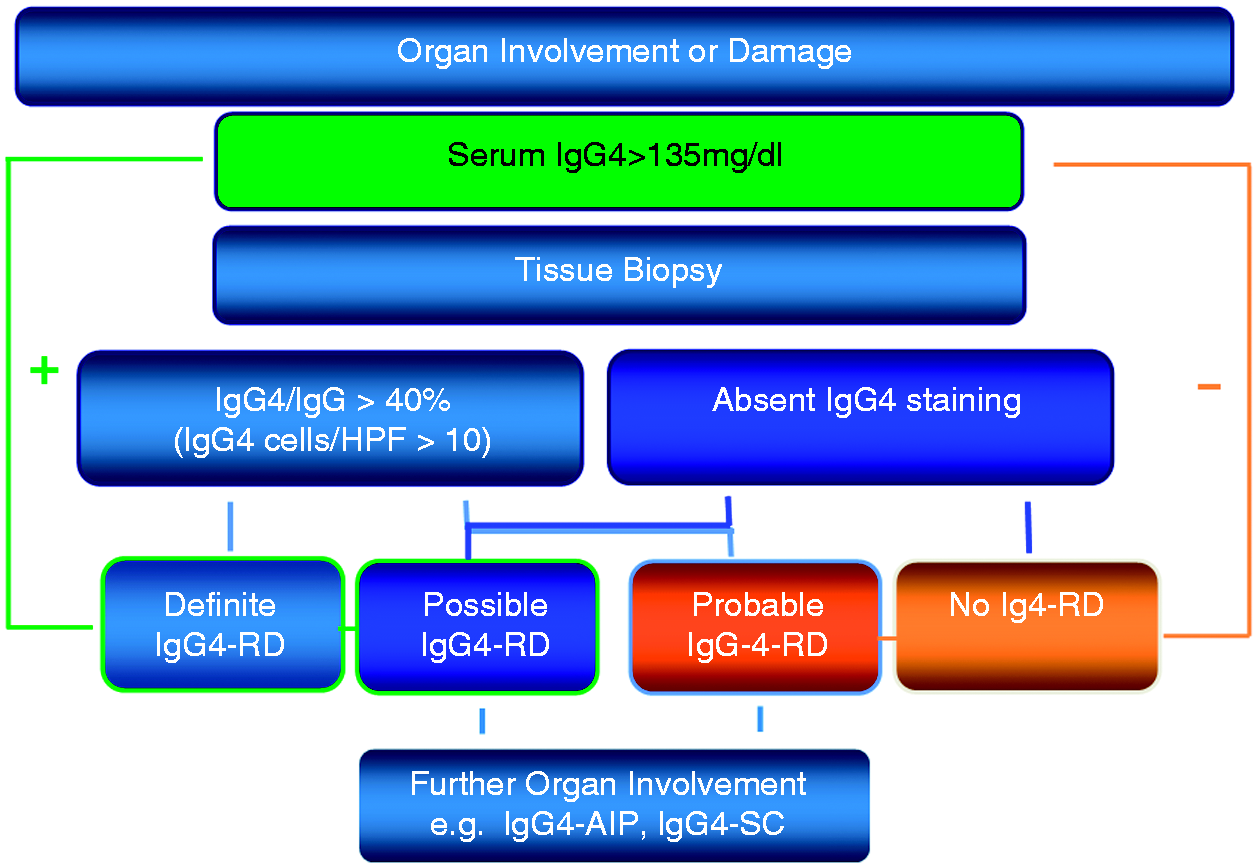
The Japanese IgG4-related disease study group has recently published a diagnostic guideline for IgG4-related disease,2,37 which allows a comprehensive approach, fully aware that the clinical presentation can be very variable depending on the site of manifestation (Table 2). The proposed guidelines include three criteria: characteristic diffuse swelling or organ mass, increased serum IgG4 levels of greater than 135 mg/dl and a positive histology. With all three criteria being positive, the definitive diagnosis can be made. With negative histology or histology not performed, the diagnosis is ‘possible IgG4-related disease’, and with positive histology but no elevated serum markers, the diagnosis is ‘probable IgG4-related disease’ (Figure 3). This classification showed satisfactory sensitivity for possible IgG4-related disease when retrospectively applied to cohorts with proven IgG4-related Mikulicz's disease, kidney disease, or AIP type 1. Thus it can be applied in clinical practice to justify further diagnostic work up when the suspicion of an IgG4-related disease arises. Organ-specific guidelines as published for Mikulicz's disease or AIP should be used in addition.2,12,37 Problems might occur due to the fact, that for the definite diagnosis histology is required which often is difficult to obtain (e.g. from the retroperitoneum, the orbita, or the pancreas). In addition, it is noteworthy that the International Consensus Diagnostic Criteria for AIP consider as diagnostic an IgG4 level more than twice the level of normal.11,12
Diagnostic algorithm for IgG4-related disease. Comprehensive diagnostic criteria for IgG4-related disease Source: Okazaki et al.
2

It is important to note that other conditions such as lymphoma, Sjögren's disease, primary sclerosing cholangitis, multicentric Castleman's disease (rare lymphoproliferative disorder unrelated to IgG4 disease), inflammatory bowel disease, and different cancers will also partially fulfil the diagnostic criteria of IgG4-related disease.2,38 Therefore diagnostic work up to exclude diseases other than IgG4-related disease is mandatory and disease-specific guidelines should be followed. Since malignant lymphoma can improve under administration of steroids, a steroid trial to establish the diagnosis of IgG4-related disease should be avoided when lymphoma is a differential diagnosis, except for AIP type 1 where a steroid trial can be necessary to confirm the diagnosis in a minority of cases, according to international guidelines.37,39 Exclusion of malignancy is generally warranted in these patients but can be difficult and can often only be achieved by histological sampling of the affected tissue.
Treatment
IgG4-related disease responds rapidly to steroid treatment with a nearly 100% initial response rate.19,20 Relapse is not a rare event (up to 47%), based on data from AIP patients.20,40 Treatment protocols vary in accordance to local standards. While groups from the USA favour a 4-week course of 40–80 mg prednisolon per day, 41 followed by steroid tapering until discontinuation, Japanese treatment guidelines for AIP recommend a 2–4-week course of prednisolon with 0.6 mg/kg bodyweight, gradual taper of 5 mg per 2 weeks, and maintenance at 2.5–5 mg for up to 3 years. 42 It turns out that maintenance treatment with low-dose steroids can decrease the risk of relapse. In a Japanese study where 82% of patients with AIP type 1 received maintenance steroid therapy, 24% of patients relapsed within 3 years, 19 whereas in 47% of patients in a study from the Mayo Clinic without steroid maintenance symptoms recurred. 40 With no controlled trial currently being available, the data suggest that maintenance steroids can effectively decrease the risk of relapse in IgG4-related AIP. In case of steroid refractory disease, contraindications for steroids, or intolerable side effects, two reports, from Khosroshahi et al. 10 and Hart et al. 41 , have recently shown that immunomodulators (azathioprine, 6-mercaptopurin, mycophenolate mofetil) and the chimeric anti-CD20-antibody rituximab are reasonable alternatives for treatment of IgG4-related disease. With no comparative clinical trial available, the decision to escalate treatment from steroids to immunomodulators or rituximab must be made on an individual case-to-case basis.
Conclusion
IgG4-related disease is an emerging clinical concept that includes a variety of known as well as new clinical entities such as Mikulicz's disease, Riedels thyroiditis, Küttner's tumour, Ormond's disease, or AIP type 1. Organ manifestations can occur simultaneously or metachroneously at multiple locations and typically include diffuse swelling or mass formation. Symptoms vary depending on the respective localization. The diagnosis is made by a combination of typical organ changes on imaging, elevated serum IgG4 levels, and histology. However, exclusion of differential diagnoses with similar clinical appearance such as malignant lymphoma, Sjögren's syndrome, cancer, and Castleman's disease is warranted. Once the diagnosis is established, steroids are the treatment of choice, which usually leads to remission.
Footnotes
Funding
This work was supported by Alfried-Krupp-von-Bohlen-und-Hahlbach-Stiftung (Graduate Schools Tumour Biology and Free Radical Biology), the Deutsche Krebshilfe/Dr Mildred-Scheel-Stiftung (109102), the Deutsche Forschungsgemeinschaft (DFG GRK840-E3/E4, MA 4115/1-2/3, NI 1297/1-1), the Federal Ministry of Education and Research (BMBF GANI-MED 03152061A and BMBF 0314107), and the European Union (EU-FP-7: EPC-TM and EU-FP7-REGPOT-2010-1).