
Abstract
Chronic erythroderma with persistent hypereosinophilia presents significant diagnostic and therapeutic challenges. We report the case of a 26-year-old man with a 6-year history of refractory erythroderma and eosinophilia. Extensive workup excluded malignancy, autoimmune disease, and secondary causes of eosinophilia. Genetic testing revealed no pathogenic variants but identified a variant of uncertain significance in HTRA2. High-dose mepolizumab monotherapy significantly reduced eosinophil counts but yielded modest clinical improvement. The addition of abrocitinib, previously ineffective alone, led to rapid and sustained symptomatic and biochemical remission. The patient experienced marked improvement in pruritus, scaling, and quality of life; subsequent withdrawal of mepolizumab led to symptom recurrence. This case highlights the complexity of managing erythroderma with hypereosinophilia and suggests that eosinophilia may be a disease marker rather than the primary symptom driver. Combined targeting of eosinophils and broader cytokine pathways via anti-interleukin-5 and JAK1 inhibition may be necessary for effective disease control in refractory cases.
Introduction
Erythroderma is a severe inflammatory skin disorder characterized by widespread erythema and scaling affecting more than 90% of the body surface area (BSA). 1 It may arise from conditions such as atopic dermatitis, psoriasis, cutaneous T-cell lymphoma (CTCL), or hypereosinophilic syndrome (HES), and often presents significant diagnostic and therapeutic challenges.1,2
HES is a hematologic disorder defined by persistent eosinophilia (>1.5 × 109/L) in the absence of identifiable secondary causes. 3 It may lead to multi-organ involvement, including frequent cutaneous manifestations. 3 Idiopathic, myeloproliferative, and lymphocytic subtypes are recognized. 3
We report a complex case of chronic erythroderma with sustained hypereosinophilia, ultimately managed successfully with combination high-dose mepolizumab and abrocitinib, following failure of multiple prior therapies—including separate trials of both agents.
Case report
A 26-year-old man was referred to our Genodermatology clinic in February 2024 with a 6-year history of chronic erythroderma, characterized by diffuse erythema, pruritus, and scaling (Figures 1–3). His medical history included childhood eczema, with no relevant family history. He reported unintentional weight loss of 12 kg in 2021, during the early course of his disease.

Clinical presentation at initial visit (February 2024), showing widespread erythema, scaling, and lichenification on the back.

Clinical presentation at initial visit (February 2024), showing widespread erythema, scaling, and lichenification on the chest.

Clinical presentation at initial visit (February 2024), showing widespread erythema, scaling, and lichenification on the lower extremities.
Prior to referral, from 2021 to 2023, he underwent extensive evaluations for progressive erythroderma. He had skin biopsies showing subacute spongiotic dermatitis with psoriasiform features, without evidence of T-cell clonality. Immunophenotyping revealed CD3⁺, CD4⁺, CD8⁺, and CD20⁺ cells with loss of CD7 and a balanced CD4:CD8 ratio. T-cell receptor gene rearrangement was negative. A transient atypical lymphocyte population appeared on the blood smear but subsequently resolved. Detailed viral serologies, autoimmune markers, and hematologic malignancy workup, including positron emission tomography scans and repeated biopsies, were negative. Notably, laboratory tests showed hypereosinophilia (3.58 × 109/L).
Over the course of this evaluation, the patient failed multiple therapies, including biologics (secukinumab, brodalumab, dupilumab, tralokinumab, spesolimab), Janus Kinase (JAK)/Tyrosine Kinase 2 inhibitors (upadacitinib, abrocitinib, deucravacitinib), immunosuppressants (methotrexate, cyclosporine), and phototherapy (Psoralen plus ultraviolet A (PUVA)). While some treatments offered transient or partial symptom relief, none resulted in sustained improvement, and the disease course remained refractory. During this period, he also experienced intermittent lymphadenopathy and severe disease flares, but no definitive evidence of CTCL or hematologic malignancy.
At referral in 2024, examination revealed violaceous, lichenified plaques with adherent scales covering 90% BSA, alopecia, and fissured fingertips (Figures 1–3). Labs confirmed persistent hypereosinophilia (3.26 × 109/L) and markedly elevated IgE (9480 IU/mL).
A comprehensive workup, including bone marrow biopsy and extensive genetic panels (600+ genes) screening for ichthyoses, primary immunodeficiencies, autoinflammatory syndromes, and eosinophilia-associated myeloid neoplasms (PDGFRA, PDGFRB, FGFR1), revealed no pathogenic variants. 4 Additional cytogenetic and molecular studies (FLT3-ID, ETV6, DDX41) were also negative.
Given these findings, a diagnosis of idiopathic HES was made. Mepolizumab (anti-interleukin (IL)-5 antibody) was initiated in July 2024. After 3 months of mepolizumab monotherapy, eosinophil counts significantly decreased (from 3.20 × 109/L to 0.52 × 109/L), but clinical improvement was modest. In November 2024, abrocitinib, a JAK1 inhibitor, was re-added adjunctively. Unlike prior treatment combination with abrocitinib–tralokinumab, when combined with mepolizumab, this regimen led to marked reduction in pruritus, scaling, and pain, with near-normalization of eosinophils (0.47 × 109/L; Figures 4 and 5). He reported significantly improved quality of life, reduced erythema, resolved flaking, and only mild pruritus (Figures 4 and 5).

Clinical improvement following combination therapy with mepolizumab and abrocitinib (September 2025). Erythema and scaling have largely resolved, with residual lichenification on the back.
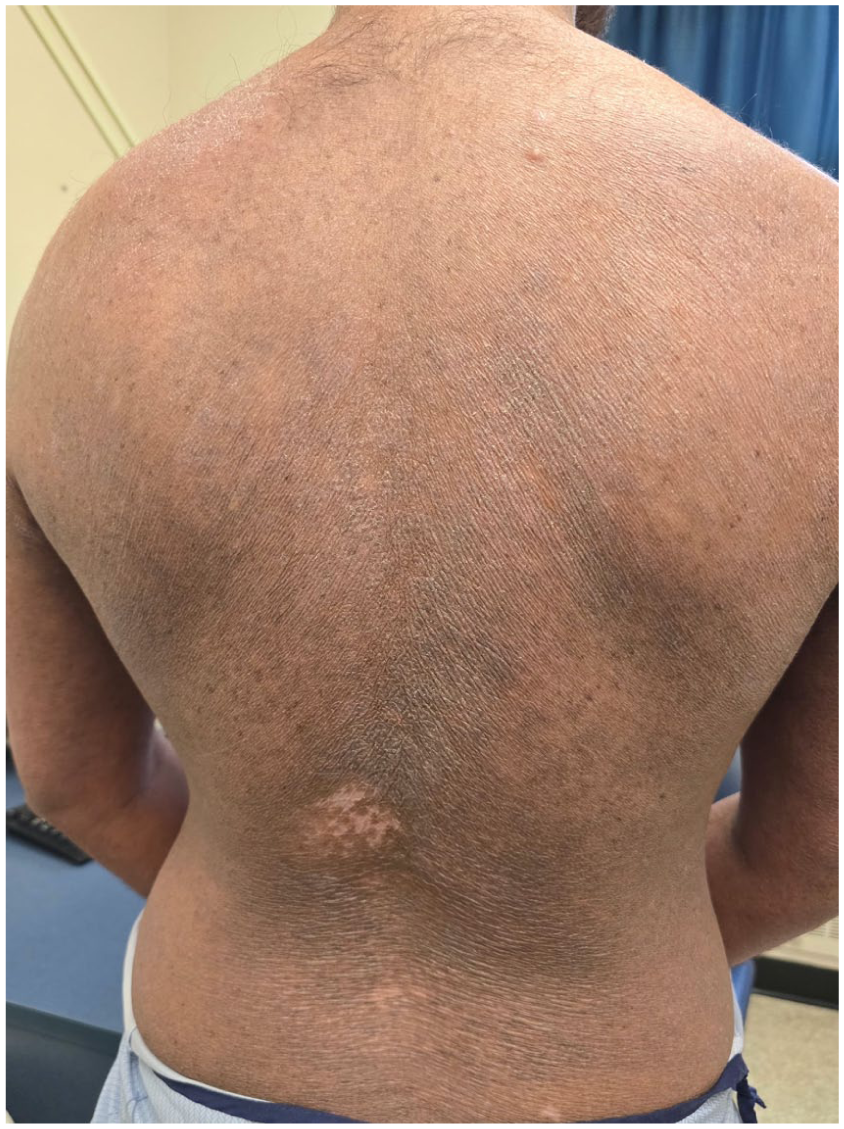
Clinical improvement following combination therapy with mepolizumab and abrocitinib (September 2025). Erythema and scaling have largely resolved, with residual lichenification, mild hyperpigmentation, and visible hair regrowth on the chest.
In April 2025, expanded genetic testing (Invitae Bone Marrow Failure Panel; 116 genes) identified a variant of uncertain significance in the HTRA2 gene (c.77G>A, p.Gly26Glu), which encodes a serine protease involved in mitochondrial function and apoptosis regulation. 5 The clinical relevance of this finding in eosinophilia remains unclear.5,6
At a visit in September 2025, the patient had stopped mepolizumab for 2 months and remained on abrocitinib monotherapy, and symptoms had begun to recur.
Discussion
This case highlights the diagnostic and therapeutic complexity of chronic erythroderma with hypereosinophilia. After excluding CTCL, autoimmune disease, and genetic syndromes, idiopathic HES was diagnosed. Persistent eosinophilia and comprehensive exclusion of secondary causes were key to this diagnosis.
Mepolizumab, an anti-IL-5 antibody, was chosen for its proven efficacy in eosinophilic disorders, including HES. 7 By targeting IL-5—a key cytokine in eosinophil activation, proliferation, and survival—mepolizumab reduces flare frequency, corticosteroid dependence, and improves symptoms.7,8 However, despite significant eosinophil reduction after 3 months of monotherapy, clinical improvement was minimal in our patient, suggesting that eosinophilia may be a marker rather than the primary driver of disease.
Incomplete responses to mepolizumab are more common in certain HES subtypes, particularly lymphocytic HES (L-HES) or multi-organ involvement, where additional immune pathways likely contribute to inflammation.7,8 JAK inhibitors such as ruxolitinib and tofacitinib have shown promise in these contexts; in a series of L-HES patients with cutaneous involvement, JAK inhibition led to rapid and sustained normalization of eosinophil counts alongside symptom control. 9
Evidence from other eosinophilic disorders supports JAK inhibitor use. Baricitinib, a JAK1/2 inhibitor, has induced glucocorticoid-free remission in chronic eosinophilic pneumonia by blocking multiple cytokine pathways (including IL-6, IL-12, IL-20, IL-22, IL-23, interferon gamma) and modulating Th2 cytokines (IL-4, IL-5, IL-13). 10 Moreover, JAK2 inhibition can reduce IL-5-independent eosinophil migration and activation, suggesting a broader immunomodulatory role. 11
Our patient’s clinical response to the combination of mepolizumab and abrocitinib—despite limited benefit from either agent used alone, and from a prior abrocitinib-tralokinumab trial—suggests either a potential synergistic effect between anti-IL-5 and JAK1 inhibition, or that eosinophils may not be the primary drivers of his symptoms. Mepolizumab monotherapy significantly reduced eosinophil counts without a meaningful clinical response, while abrocitinib alone was previously ineffective. However, the combination led to marked symptomatic and biochemical improvement. This outcome raises the possibility that eosinophilia may function more as a disease marker than a direct driver of symptoms, and that combined targeting of eosinophils and broader cytokine pathways may be necessary for effective disease control. Furthermore, withdrawal of mepolizumab led to the recurrence of symptoms.
In conclusion, this case highlights the importance of a multidisciplinary approach to managing complex, refractory erythroderma, particularly in younger patients with systemic involvement. The combination of mepolizumab and abrocitinib may offer a promising strategy for managing eosinophilic dermatoses unresponsive to conventional and monotherapies, emphasizing the need for personalized therapeutic approaches.
Footnotes
Consent to participate
M.F. reports serving on advisory boards for Celltrion, Sanofi, Medison Pharma and Bausch Health, honoraria from GSK, Novartis, Stallergenes and Medexus, and speaking fees from Bausch Health. Investigator funding from Cogent. MBS has served as a speaker, consultant, investigator, or investigator for Celltrion Medexus, Novartis Pharmaceuticals, Pfizer, Sanofi Genzyme and ALK . MBS received honoraria, consulting fees, and principal investigator funding. EN has served as a speaker, consultant, investigator, or advisory board member for AbbVie, Apogee, Arcutis, Bausch Health, BioJamp, Boehringer Ingelheim International, Bristol Myers Squibb, Celltrion, Eli Lilly, Galderma, Innovaderm, Janssen, LEO Pharma, Medexus, Novartis Pharmaceuticals, Organon, Pfizer, Sanofi Genzyme, Searchlight Pharma, Sun Pharmaceuticals, and UCB. EN has received honoraria, consulting fees, and investigator-initiated research funding.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding support from Canadian Dermatology Foundation Grant.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.