
Abstract
Pulmonary embolism in the pediatric population is rare but could be a cause of significant morbidity and mortality. Pleural effusion often complicates pulmonary embolism but it is usually small to moderate in size. We report the case of a 16-year-old male patient with massive pleural effusion as a delayed complication of pulmonary embolism. He presented with pleuritic chest pain, dyspnea, and deep venous thrombosis in the leg, the risk factors for which were obesity and a hypercoagulable state due to a factor V Leiden heterozygous mutation. The patient received anticoagulation treatment with enoxaparin. Three weeks after discharge home, he was readmitted for new-onset chest pain and dyspnea. He was found to have massive left pleural effusion without recurrent pulmonary embolism. The effusion was drained because it compromised the patient’s respiratory function. The mechanism for pleural effusion in pulmonary embolism is unclear but may involve increased pulmonary capillary permeability due to inflammatory mediators released from platelet-rich thrombi and pulmonary ischemia distal to the vascular obstruction. This case highlights the potential for late pleural effusion progression in pediatric pulmonary embolism and the need to manage large effusions to prevent respiratory compromise.
Keywords
Introduction
Although pulmonary embolism (PE) is common in adults, it is relatively rare in the pediatric population. 1 Data from the National Hospital Discharge Survey in the United States from 1979 to 2001 showed annual incidence rates of 0.9 PE cases/100,000 children and 4.9 venous thromboembolism cases/100,000 children with the highest incidence rate in infants <1 year of age and a second peak in adolescents. 2 A recent retrospective study, which analyzed data from the Kids’ Inpatient Database in the United States for the years 2016 and 2019, reported a higher annual incidence rate of pediatric PE of 3.5 cases/100,000 people. This study similarly identified two peaks in incidence rates during infancy and adolescence. 3 The incidence of pediatric PE has been increasing steadily, which can be attributed to several factors, including increased awareness and recognition, increased survival of pediatric patients with underlying predisposing conditions, increased use of central venous catheters in newborns and infants, hormonal supplementation in female adolescents, and the availability of noninvasive diagnostic modalities. 4 An overall in-hospital case fatality rate of 4.5% was reported in pediatric PE and up to 25.3% in patients with high-risk features. 3 PE as a cause of pleural effusion is a commonly overlooked etiology, even though pleural effusion occurs in 30%–50% of patients with PE. 5 Most pleural effusions due to PE are small, usually causing only blunting of the costophrenic angles. 5 Massive pleural effusion in PE is uncommon, particularly as a delayed presentation. In a large retrospective study of PE in adult patients, 95% of the effusions occupied less than a third of the hemithorax. 6 The association between PE and pleural effusion in the pediatric population is not well documented. To our knowledge, no previous case of delayed massive pleural effusion in pediatric PE has been reported in the literature. In one case series of five adult patients with delayed diagnosis of PE, pleural effusions were found to be loculated and resolved with anticoagulant therapy and without the need for drainage. 7 We present a case report that describes a unique presentation of massive pleural effusion occurring several weeks after the initial diagnosis and treatment of PE in an adolescent patient. This case highlights the importance of continued vigilance in the follow-up of pediatric PE patients, even after initial improvement. The timeline of this case report is presented chronologically in Table 1.
Timeline of major clinical events and hospital course.

CTPA: computed tomography pulmonary angiography; DVT: deep venous thrombosis; ED: Emergency Department; LMWH: low-molecular-weight heparin; PE: pulmonary embolism; PICU: Pediatric Intensive Care Unit.
Case Narrative
Presenting concerns
A 16-year-old Caucasian male presented to the Emergency Department (ED) with a history of sudden onset dyspnea and bilateral pleuritic chest pain that commenced a few hours prior. The patient had been diagnosed with deep venous thrombosis (DVT) of the left leg 3 weeks earlier. He was undergoing treatment with apixaban and was being monitored by the hematology/oncology team on an outpatient basis. The patient had discontinued apixaban use for 2 days preceding the current presentation due to concerns about bloody stools.
Clinical findings
The patient’s vital signs upon initial physical examination were as follows: temperature, 38.3°C; respiratory rate, 26 breaths/min; heart rate, 126 beats/min; blood pressure, 146/105 mmHg; and oxygen saturation, 96% on room air. The patient had a body mass index of 49.5 kg/m2 (morbid obesity). The patient did not exhibit any signs of respiratory distress, and his chest was clear on auscultation. Shortly after, he was noted to have a low oxygen saturation of 92% on room air; so, he was placed on oxygen via a Venturi mask at 9 L/min. Mild edema in the left leg was observed with no calf tenderness. The rest of his physical examination was unremarkable.
Diagnostic assessment
Considering the recent history of DVT, there was a significant suspicion of PE. Computed tomography pulmonary angiography (CTPA) was promptly performed, revealing findings that were highly indicative of PE. The CTPA demonstrated filling defects extending distally in the left lower interlobar pulmonary artery (Figure 1(a)) and the segmental branches of the pulmonary artery in the right upper and middle lobes (Figure 1(b)), accompanied by bilateral pulmonary parenchymal ischemic changes and infarcts. No pleural abnormality was present. The initial laboratory workup, detailed in Table 2, indicated thrombosis, as evidenced by an elevated D-dimer level of 1.65 µg/mL. Furthermore, the fibrinogen level was elevated to 593 mg/dL. Venous blood gas analysis revealed a mild respiratory alkalosis. The complete blood count (CBC) and complete metabolic panel results were within normal limits. A venous duplex ultrasound study of both lower limbs identified acute DVT in the left leg, involving the femoral, popliteal, posterior tibial, and peroneal veins.

(a) Axial CTPA section shows a filling defect in the left lower interlobar pulmonary artery. (b) Axial CTPA section shows filling defects in the right and left segmental branches of the pulmonary artery.
Pertinent initial laboratory parameters.
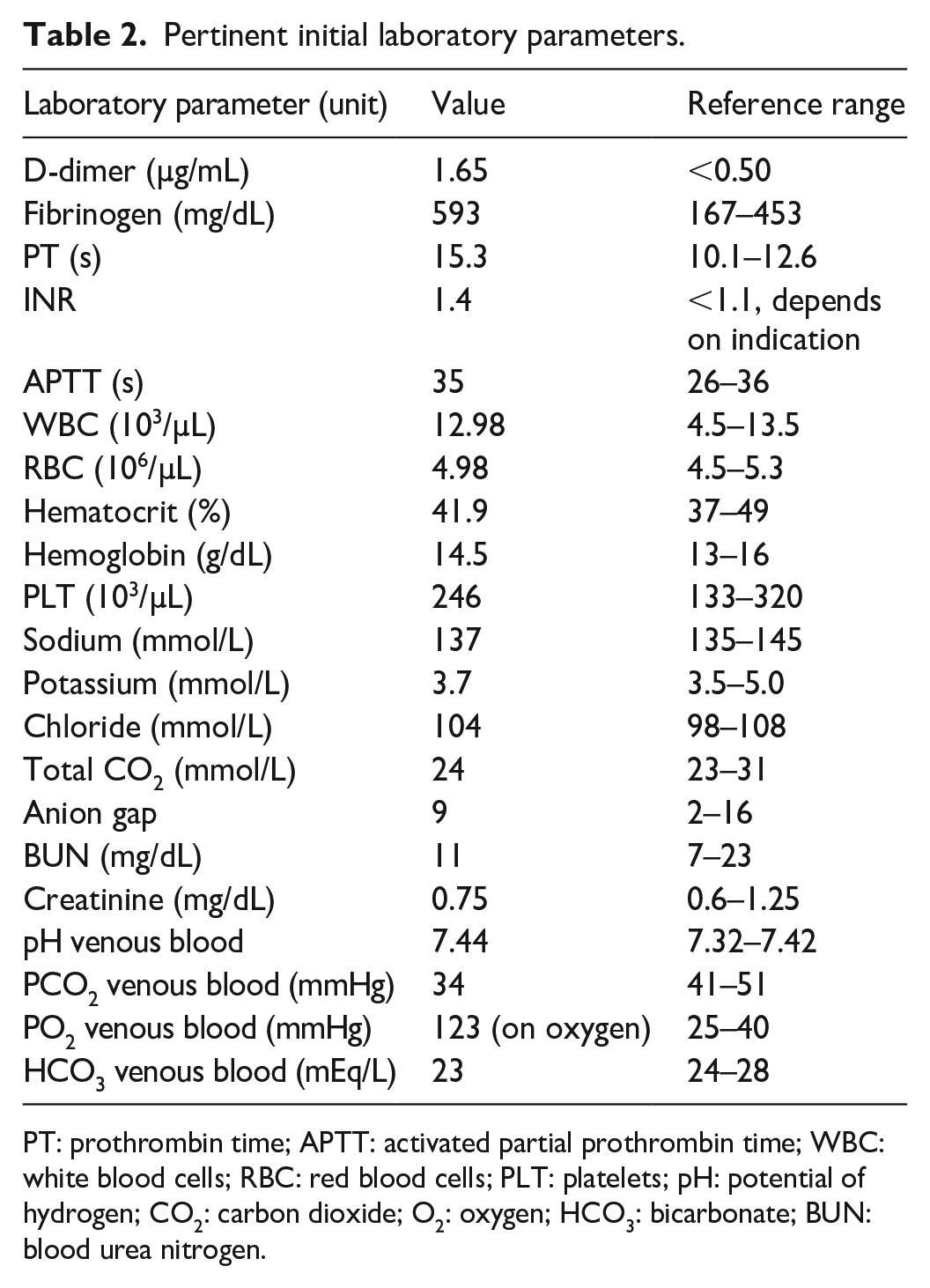
PT: prothrombin time; APTT: activated partial prothrombin time; WBC: white blood cells; RBC: red blood cells; PLT: platelets; pH: potential of hydrogen; CO2: carbon dioxide; O2: oxygen; HCO3: bicarbonate; BUN: blood urea nitrogen.
Therapeutic intervention
The patient was admitted to the Pediatric Intensive Care Unit (PICU) and commenced on anticoagulation therapy with enoxaparin, initially administered at a dose of 100 mg subcutaneously every 12 h. The therapeutic effect was assessed following the third dose by measuring anti-Xa low-molecular-weight heparin (LMWH) levels, which were monitored daily. The enoxaparin dosage was adjusted accordingly until a therapeutic effect was achieved at a dose of 150 mg administered subcutaneously every 12 h. In addition, the patient received supportive care including analgesia, intravenous fluids, stress ulcer prophylaxis, and supplemental oxygen.
Follow-up and outcomes
The hematology/oncology team participated in the management of the patient. A comprehensive thrombophilia evaluation revealed factor V Leiden heterozygous mutation. A chest X-ray (Figure 2) was performed on day 2 of hospitalization due to the presence of fevers. The maximum temperature was 39.5°C. The chest X-ray showed bibasilar opacities. In response to a potential pneumonia diagnosis, the patient received intravenous ceftriaxone at a dosage of 2 g daily for 48 h, followed by oral cefdinir (300 mg daily) to complete a 7-day treatment regimen. CBC was normal without leukocytosis (white blood cell (WBC) count was 11.48 × 103/µL). Blood culture yielded no growth. Given the persistent fevers, follow-up CTPA (Figure 3) was performed on day 3 of hospitalization. The CTPA revealed unchanged PE, an extension of pulmonary infarcts, a trace right pleural effusion, and a small left pleural effusion, which did not further compromise the patient’s respiratory function. Echocardiography showed no evidence of right heart strain. By day 4 of hospitalization, therapeutic levels of anti-Xa LMWH were achieved. The patient demonstrated clinical improvement over the next few days. Chest X-ray on day 7 of hospitalization showed a stable, small, left pleural effusion (Figure 4). However, attempts to completely discontinue oxygen therapy were unsuccessful due to an oxygen saturation level of 89% on room air. The prolonged dependence on oxygen was attributed to a ventilation-perfusion mismatch. On day 9 of hospitalization, the patient was discharged home with a prescription for enoxaparin at 150 mg every 12 h and oxygen therapy at 2 L/min via nasal cannula.
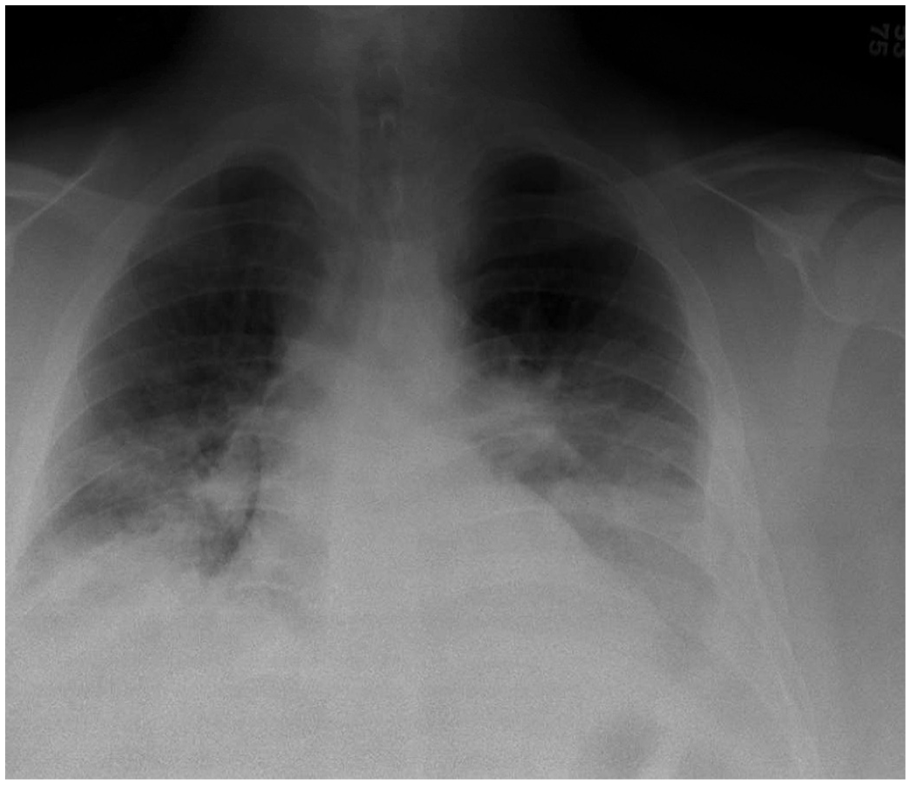
Chest X-ray on day 2 of hospitalization shows low lung volume and bibasilar opacities.

Coronal CTPA section on day 3 of hospitalization shows a left pulmonary infarct and small left pleural effusion.

Chest X-ray on day 7 of hospitalization shows low lung volume and a stable small left pleural effusion.
Following his discharge, the patient attended two outpatient follow-up visits at the hematology/oncology clinic. During these evaluations, he was determined to be in stable condition and had discontinued the use of oxygen therapy. However, 3 weeks post-discharge, the patient returned to the ED, presenting with newly developed dyspnea and left-sided chest pain. The patient reported the onset of dyspnea and left-sided pleuritic chest pain upon awakening from sleep, ~2 h before the presentation. Physical examination revealed an afebrile patient with a temperature of 37.5°C who was tachypneic with a respiratory rate of 25 breaths/min, heart rate of 108 beats/min, a blood pressure of 128/83 mmHg, and oxygen saturation of 95% on room air. Auscultation indicated reduced air entry in the left lung zone. Chest X-ray (Figure 5) revealed massive left pleural effusion. CTPA (Figure 6) demonstrated massive left pleural effusion and no evidence of PE. Laboratory evaluation did not suggest an infectious process. The patient was afebrile and had a normal WBC count of 10.97 × 103/µL. A normal echocardiogram and N-terminal pro-B-type natriuretic peptide level of 46 pg/mL (reference value ⩽125 pg/mL) made a cardiac cause for the pleural effusion unlikely. Subsequently, the patient required oxygen supplementation at a rate of 1 L/min due to a decrease in oxygen saturation to 88%–90% while breathing room air. The patient was readmitted to the PICU. Enoxaparin was discontinued, and a continuous infusion of heparin was initiated at a rate of 18 units/kg/h. The therapeutic effects were assessed every 4 h using anti-Xa unfractionated heparin levels. On the day after admission, a consultation with the pulmonology team was conducted, during which bedside thoracentesis was performed, yielding 1400 mL of serosanguineous fluid. They subsequently recommended the placement of a chest drain while continuing anticoagulation therapy. Two hours before insertion of the chest tube, heparin infusion was discontinued, and enoxaparin was resumed following the procedure. A 10-French chest tube was inserted into the left pleural space under ultrasound guidance. During the procedure, an additional 1400 mL of serosanguineous fluid was drained. The chest tube was subsequently connected to a closed drainage system, facilitating the further evacuation of 1200 mL of pleural effusion over 24 h. An additional 80 mL of pleural fluid was evacuated the following day before the chest tube was clamped. The tube was removed 2 days after insertion. In total, 4080 mL of serosanguineous fluid was evacuated from the left pleural space over 2 days. Pleural fluid analysis revealed a glucose level of 106 g/dL, lactate dehydrogenase level of 265 U/L, and protein level of 5.5 g/dL with a pleural fluid/serum protein ratio of 0.74, consistent with an exudate. The culture did not yield any growth, and cytology revealed no malignant cells. The patient received chest physiotherapy and utilized incentive spirometry to facilitate lung re-expansion. During this hospitalization, the patient did not receive antibiotic therapy. The patient recovered well and was discharged after a 4-day hospital stay to continue enoxaparin at home. He transitioned to warfarin after 4 weeks of enoxaparin use while continuing enoxaparin for 1 additional week. He is currently being followed up as an outpatient by the hematology/oncology team and receiving long-term anticoagulation therapy with warfarin. He remains in good general health.
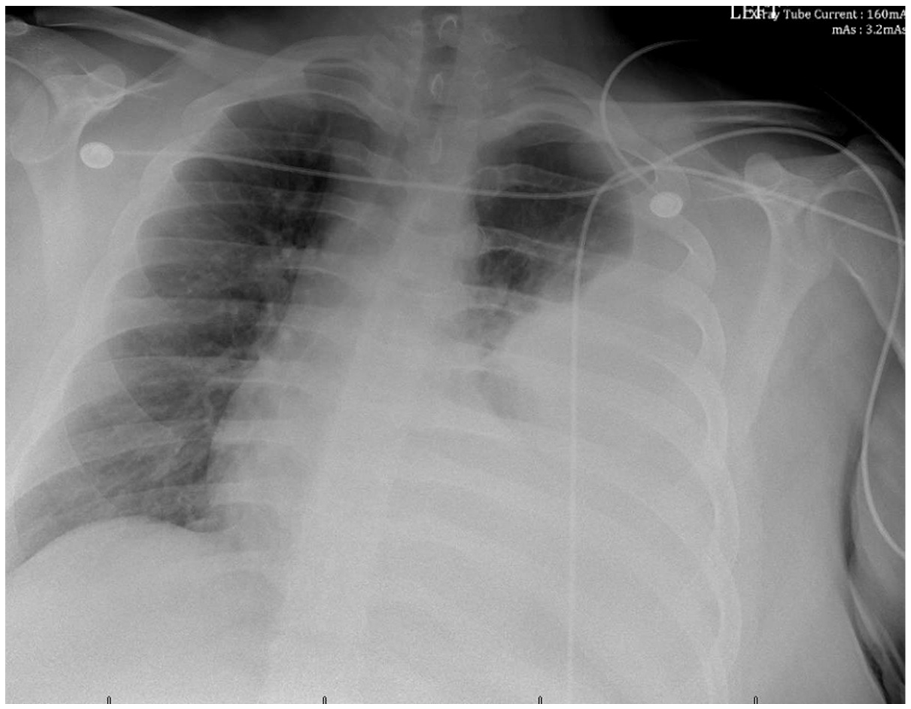
Chest X-ray 1 month after PE shows massive left pleural effusion and left lung atelectasis.

Coronal CTPA section 1 month after PE shows massive left pleural effusion with near complete collapse of the left lung and a rightward mediastinal shift.
Discussion
This case illustrates the potential for delayed progression of pleural effusion in pediatric PE. The pathogenesis of pleural effusion in PE remains inadequately understood. Pleural effusions due to PE are usually exudates, believed to occur due to increased permeability of the pulmonary capillaries. The mechanism by which this occurs has been postulated to be due to the release of inflammatory mediators, notably vascular endothelial growth factor (VEGF), from platelet-rich thrombi. VEGF is a potent mediator of increased vascular permeability. Elevated levels of VEGF have been documented in pleural effusion associated with PE. The resulting excessive interstitial lung fluid traverses the visceral pleura and accumulates in the pleural space as pleural effusion.5,8,9 Ischemia of the pulmonary capillaries beyond the site of the embolus may also contribute to increased capillary permeability. Visceral pleural ischemia is unlikely to contribute to the formation of pleural effusion due to its dual blood supply from the bronchial and pulmonary circulation. 5 Hypoxia and ischemia increase the cellular expression of VEGF, contributing to the increased elaboration of VEGF in the pleural fluid in PE. 9 Pleural fluid accumulation occurs due to alterations in the balance between fluid formation and resorption in the pleural space. 10 There is no correlation between the sidedness of the pleural effusion with that of the PE. 11 Most PE-associated pleural effusions are small and unilateral.8,11,12 They occur and reach maximal size early during the PE. 12 In a study by Bynum and Wilson, maximal pleural effusion was observed within 3 days of the PE among 60 out of 62 patients studied. 12 Late enlargement of pleural effusion in PE suggests a complication such as hemothorax or recurrent PE or pleural infection.8,12 Delayed pleural effusion following PE may also be attributed to Dressler’s syndrome, a rare complication that can manifest days to weeks after PE. This syndrome is characterized by fever, pericarditis, and leukocytosis with or without accompanying pleural effusion. 8 While pleural effusions associated with PE are typically free-flowing, they may become loculated if the diagnosis of PE is delayed, as documented in previous studies.7,11 Bynum and Wilson found that pulmonary infarction was associated with longer-lasting and larger pulmonary effusions. 12 In contrast, Porcel et al. reported smaller pleural effusions in cases of pulmonary infarction. Furthermore, their study did not observe an increased incidence of pleural effusions in patients with pulmonary infarction compared to those without. The authors proposed that a substantial volume of pleural fluid leading to passive atelectasis might obscure the detection of certain parenchymal opacities. In addition, they propounded that ischemia may induce mesothelial cells to secrete various coagulation cascade proteins, such as tissue factor, resulting in procoagulant activity and fibrin deposition in the pleural space, leading to spontaneous pleurodesis, preventing the accumulation of pleural fluid. 11
The cause of the delayed progression of pleural effusion in our patient over the course of 1 month remains unclear. Pleural infection is unlikely to be the cause of this late progression, as the patient did not exhibit leukocytosis and remained afebrile during the second hospitalization. Although the patient experienced fever during the first hospitalization, the presence of fever alone could not reliably distinguish acute PE from PE complicated by pneumonia. Numerous studies have documented fever incidence ranging from 14% to 57.1%, attributed solely to acute PE.13–15 Patients with acute PE may present with fever, including high-grade fever. It is crucial to consider the possibility of PE and not dismiss it in favor of other conditions, such as pneumonia. 14 Although the patient received empirical antibiotic therapy, a diagnosis of pneumonia was not confirmed. Performing diagnostic thoracentesis before the initiation of antibiotic therapy may aid in distinguishing between pleural effusion caused solely by PE and complicated parapneumonic effusion. However, the small sizes of most PE-associated pleural effusions often preclude this. The finding of a pleural fluid glucose level >60 mg/dL in our patient further buttresses the point that the delayed expansion of the effusion was not likely due to an infection. A pleural fluid pH below 7.20 or a glucose level below 60 mg/dL generally indicates a complicated parapneumonic effusion and a need for drainage.10,16 Other potential causes of delayed pleural effusion in PE, such as hemothorax and recurrent PE, were ruled out. The patient’s presentation was not consistent with Dressler’s syndrome. One plausible explanation for the patient’s clinical presentation is that the extensive pulmonary infarction elicited a robust and prolonged inflammatory response, accompanied by a sustained increase in pulmonary capillary permeability, which subsequently resulted in delayed massive pleural effusion. This presentation likely represents one form of the natural history of pleural effusion caused by PE.
Conclusion
This case highlights the importance of continued monitoring and follow-up in pediatric patients with PE, even after initial improvement. Important red flags that may indicate late progression of PE-associated pleural effusion include recurrence or worsening of respiratory symptoms, such as dyspnea and chest pain. It is important to consider PE in the differential diagnosis of all cases of pleural effusion in the pediatric population. Patients may be screened by D-dimer testing, and if positive, further testing with CTPA is recommended. While most effusions in PE resolve with anticoagulation alone, large and massive effusions must be drained to prevent respiratory compromise. Further research is needed to better understand the pathogenesis of pleural effusion in PE, as well as the factors contributing to its delayed onset and late progression in the pediatric population.
Footnotes
Acknowledgements
The authors thank all the medical staff who were involved in the management of this patient.
Ethical considerations
Our institution does not require ethical approval for reporting individual cases or case series.
Consent for publication
Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.
Author contributions
S.A.A.-I. conducted the literature search and drafted the original manuscript. A.M. treated the patient. S.A.A.-I. and A.M. contributed to the concept, design, definition of intellectual content, and manuscript revision for the article, approved the final version to be published, and agreed to be accountable for all aspects of the work.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.