
Abstract
Autoimmune hemolytic anemia is when autoantibodies attack red blood cells, leading to anemia. Cold agglutinin disease, a subtype of autoimmune hemolytic anemia, accounts for 13%–32% of cases. We present a middle-aged woman with a history of Raynaud’s phenomenon who was admitted to the hospital due to acrocyanosis, weakness, and lethargy. Her blood tests revealed indirect hyperbilirubinemia, severe anemia, an increase in lactate dehydrogenase, a positive direct Coombs test, increased inflammatory factors, and a positive cold agglutinin antibody, confirming the diagnosis of cold agglutinin disease. Despite a 5-day hospital stay and initiation of treatments, she did not show a satisfactory response to the treatments. Cold agglutinin disease is a chronic disease that can initially present with mild symptoms. Early identification through appropriate laboratory tests is crucial. In severe cases, prompt initiation of proper treatment is necessary to prevent fatal outcomes.
Introduction
Autoimmune hemolytic anemia (AIHA) is a disease in which autoantibodies against red blood cells (RBCs) cause intravascular hemolysis, leading to anemia. A specific type of this condition is cold agglutinin disease (CAD), accounting for 13%–32% of AIHA cases. In CAD, the body produces immunoglobulin M (IgM) that recognizes antigens on RBCs and responds to them at low temperatures (below 37°C), causing hemolysis. In addition, the classical complement pathway is activated in this disease, contributing to the destruction of RBCs. Symptoms of CAD include severe anemia, livedo reticularis, acrocyanosis, Raynaud’s phenomenon, and fatigue. In America, the 5-year prevalence of this disease ranges from 14 to 33 cases/million, classifying it as a rare disease. CAD has a higher prevalence in women than men and is more common in individuals over 50 years old.1,2 These findings underscore the imperative for the timely diagnosis and treatment of these patients to enhance their prognosis.
Case
Our patient was a 57-year-old Caucasian woman who was referred to the hospital due to lethargy and jaundice. The patient’s symptoms first started 2 weeks before, on February 25, 2024, with scleral icterus along with darkening of the urine. Later, fatigue, lethargy, and permanent cyanosis of the extremities and tip of the nose were gradually added to the patient’s symptoms. She was referred to the hospital after presenting the results of laboratory tests to a doctor, which showed severe normochromic normocytic anemia (Hb = 3.5 g/dl) and evidence of hemoly-sis (increased lactate dehydrogenase (LDH) levels (LDH = 1530), positive direct Coombs, and indirect hyperbilirubinemia). She mentioned a history of Raynaud’s phenomenon from 2 years ago, which presented as intermittent pallor, cyanosis, and redness of extremities following contact with cold temperature or stress, but no further investigations were done regarding the cause of this phenomenon. The patient had no history of drug use and a family history of diseases. She had a pale and icteric appearance in the examination, and her conjunctivae were pale. The patient’s vital signs included blood pressure of 120/80 mm Hg, body temperature of 37°C, heart rate of 102 beats/min, respiratory rate of 18/min, and oxygen saturation (SpO2) of 94%. Evidence of blue discoloration and coldness in the tip of the nose and the extremities, as well as livedo reticularis, was present in all four limbs (Figure 1). The abdomen was soft and non-tender, and there was no evidence of hepatosplenomegaly.
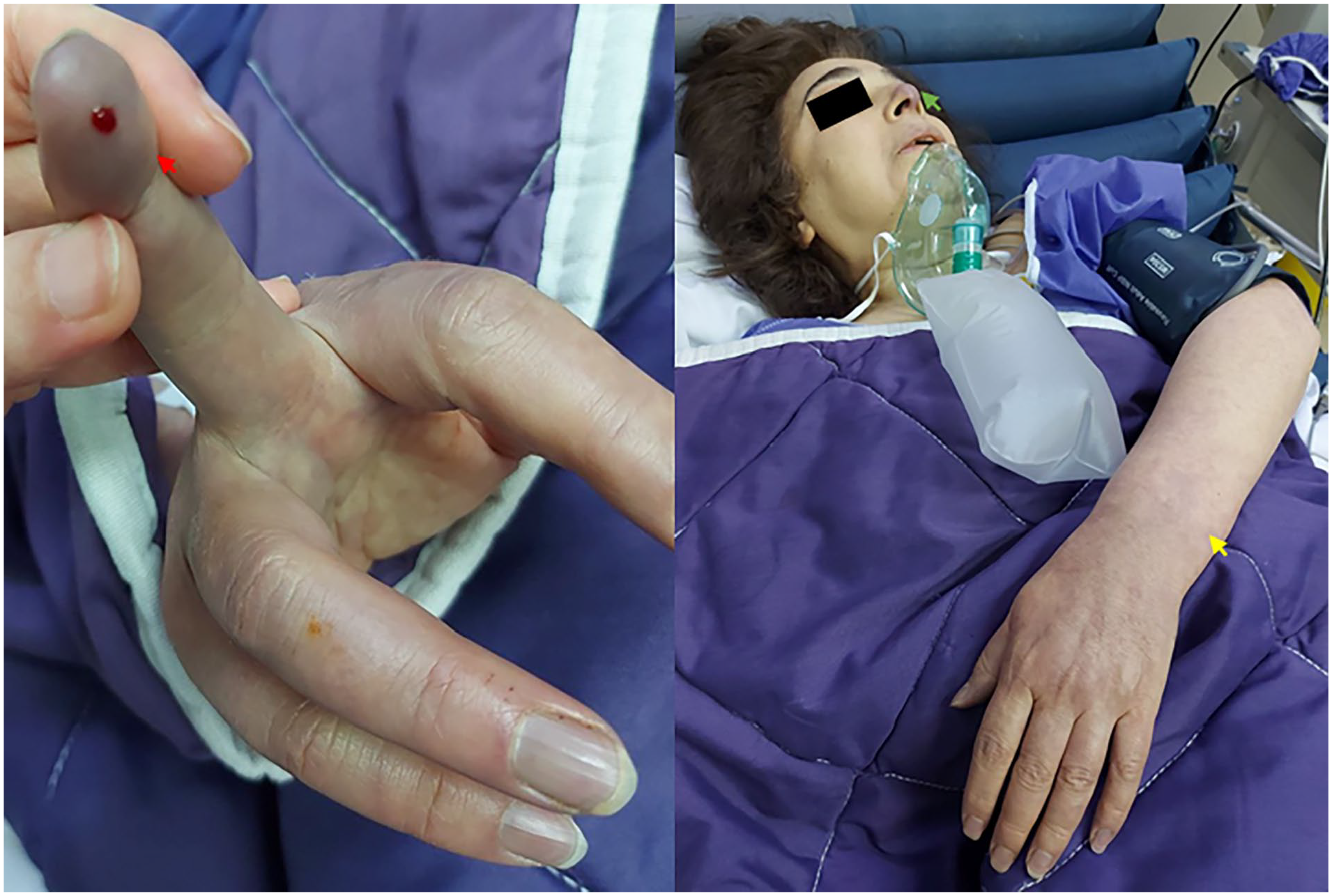
The patient presents with symptoms including pale and icteric skin, acrocyanosis (red arrow), cyanosis of the nose tip (green arrow), and livedo reticularis (yellow arrow).
Based on the results of blood tests, the patient was diagnosed with AIHA and was admitted to the intensive care unit. Further tests were conducted to determine the cause, including testing for viral infections such as respiratory syncytial virus, COVID-19, Ebstein-barr virus, influenza A, B, and H1N1, hepatitis B and C. In addition, specific tests were conducted to check for connective tissue diseases such as systemic lupus erythematosus, rheumatoid arthritis, and antiphospholipid syndrome. The immunoglobulin levels and CA antibody titer were also measured (Table 1).
Laboratory findings.

WBC: White blood cells; MCV: Mean corpuscular volume; MCH: Mean corpuscular hemoglobin; LDH: Lactate dehydrogenase; CPK; Creatinine phosphor kinase; Cr: Creatinine; CRP: C-reactive protein; ESR: Erythrocyte sedimentation rate; INR: International normalized ratio; PTT: Partial thromboplastin time; AST: Aspartate aminotransferase; ALT: Alanine aminotransferase; ABG: Arterial blood gas.
Abdominal ultrasound showed a normal-sized liver with grade 1 fatty liver, a normal-sized spleen, and no other abnormalities. RBC agglutination was observed in different fields in the patient’s peripheral blood smear (Figure 2). Electrophoresis of serum and urine proteins was normal. Her bone marrow biopsy revealed hypercellular bone marrow and erythroid hyperplasia, with a myeloid to erythroid ratio of 1:3, suggesting no preference for malignant secondary etiologies (Figure 3). In the requested urine test, the patient’s urobilinogen was +2, and the rest of the lab test results were normal (Table 1).

The patient’s peripheral blood smear showed red blood cell agglutination (orange arrow).

The patient’s bone marrow biopsy demonstrates hypercellular bone marrow and erythroid hyperplasia, with a myeloid to erythroid ratio of 1:3.
Her treatment started with 1 mg/kg of methylprednisolone for 3 consecutive days, along with intravenous immunoglobulin at a dose of 25 mg/kg for 5 days. However, her condition deteriorated, and her anemia worsened (Hb = 2.8). In addition, she started experiencing dyspnea, and her SpO2 decreased to 88%. A chest radiography was performed, but it showed no abnormality that could explain the drop in SpO2 levels. The patient’s difficulty in breathing was attributed to severe anemia, and one unit of warmed iso-group and cross-matched packed RBCs was transfused. Unfortunately, after receiving a warmed erythrocyte transfusion, the patient experienced generalized hemolysis, and there was no improvement in symptoms.
On the third day of hospitalization, the results of tests were acquired, and tests for connective tissue diseases all came back negative. IgM and IgG levels were normal, but the CA antibody titer at 4°C was 1:1280 (normal range <1:64), confirming the CAD diagnosis. As the patient did not seem to tolerate a multidrug regimen, monotherapy with rituximab was initiated for the patient with a 375 mg/m2 dose on day 1, and the plan was to continue the therapy every week for 4–8 weeks.
However, despite starting the treatment, her shortness of breath worsened, and on the fifth day of hospitalization, she experienced a cardiorespiratory arrest and unfortunately passed away.
Discussion/conclusion
Hemolytic anemia occurs when the hemoglobin decreases due to the destruction of RBCs and increased hemoglobin catabolism. This leads to increased efforts by the bone marrow to compensate for the loss of hemoglobin. These conditions increase the number of reticulocytes, LDH levels, unconjugated bilirubin, urinary urobilinogen, and decrease in haptoglobin levels. 3 As hemolysis has a wide range of etiologies, specific tests are required to help determine the underlying cause. For example, a positive direct Coombs test confirms the immunologic nature of the hemolysis and can also be positive in CAD disease. 4
In CAD, IgMs, which are also referred to as CA, react with RBCs, leading to intravascular and extravascular hemolysis. Extravascular hemolysis is associated with complement activation and phagocytosis of RBCs, mainly in the liver. Intravascular hemolysis also results from the formation of the membrane attack complex, which consists of C5b, C6, C7, C8, and C9. 5 In this case, the initial symptoms and evidence indicated the occurrence of intravascular hemolysis. Also, extensive agglutination of RBCs in the peripheral blood sample suggested the presence of high levels of CA in the peripheral blood.
After diagnosing CAD, it is essential to provide appropriate treatment based on the severity of the condition. Studies on the relationship between seasons and CAD mortality show that the highest number of deaths occurs in the winter, and symptoms during the cold season are directly related to the severity of the disease. 6 However, avoiding exposure to cold temperatures has only been effective in very mild cases of the disease and not in moderate-to-severe cases. In the context of moderate-to-severe cases, rituximab is among the treatment options utilized. However, its efficacy has been observed in only 57% of cases, with a complete response evident in merely 21%. Another drawback of rituximab treatment is its slow response time in patients with CAD. 7 Among various treatment methods, according to a newly published study conducted by Barcellini et al., rituximab still represents the frontline approach in patients with symptomatic anemia or disabling cold-induced peripheral symptoms and is effective in 50%–60% of cases. 8 In this method, treatment aims to target the pathogenic B-cell clone in the bone marrow to reduce the production of monoclonal CA. Rituximab combined with fludarabine has been linked to a 76% response rate. 9 Rituximab combination with bendamustine is also used, especially in Waldenström macroglobulinemia-associated CAD. 10 Bortezomib is also an efficient therapy in cases where rituximab is either ineffective or contraindicated. 11 One of the other treatment methods is anti-complement therapies, which reduce the need for blood transfusion by improving hemoglobin levels, rapid cessation of hemolysis, and alleviating fatigue. Sutimlimab (anti-C1s) and pegcetacoplan (anti-C3) are two drugs categorized as anti-complement therapies that are effective in patients with CAD.
Although still used as a clinical modality, corticosteroids are no longer recommended for CAD treatment, as patient response has been limited and seen only at very high doses. 12 In this case, the patient sought medical help at a late stage of the disease and experienced severe symptoms. High doses of corticosteroids were administered until a definitive diagnosis was made. After the diagnosis was confirmed, considering no detectable bone marrow lymphoproliferative disease and the possibility of not tolerating multidrug treatments, and limited accessibility of the hospitalized center for accessing treatment methods such as fludarabine, monotherapy with rituximab was started.
One notable thing about this patient is the presence of diffuse jaundice, which is an unusual finding in patients with CAD. Other factors indicating the severity of the disease in this patient include elevated levels of LDH and indirect bilirubinemia, contributing to a poor prognosis. 13
One of the limitations of this case was the late administration of rituximab, given the AIHA diagnosis. However, the patient was initially admitted to another hospital and was referred to our facility on the second day of admission. Furthermore, it took ~24 h for the patient’s sibling to procure the necessary medication, resulting in a delayed infusion. Nonetheless, given the patient’s severe condition and the delayed referral to our hospital, it is improbable that this factor would significantly alter the course of treatment or the patient’s overall condition.
The introduced patient was a CAD case without a previously diagnosed disease and only with a history of Raynaud’s phenomenon from 2 years ago, with evidence of severe disease, including acrocyanosis, lethargy, indirect hyperbilirubinemia, severe anemia, high LDH levels, positive direct Coombs and increased inflammatory factors and CA antibodies. During 5 days of hospitalization, exhaustive investigations were conducted to determine the etiology of the illness, yet no specific cause was identified. Despite the initiation of therapy, the patient did not exhibit a positive response.
CAD is a chronic condition that can start with mild symptoms but lead to fatal complications, so early diagnosis is essential.
Footnotes
Ethical considerations
Ethical approval was not sought for the present study because our institution does not require ethical approval for reporting individual cases.
Consent to participate
Written informed consent was obtained from legally authorized representatives before the study.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.