
Abstract
Mycosis fungoides is the most common cutaneous T-cell lymphoma, with its folliculotropic variant being more aggressive and often misdiagnosed. We present a case of a 38-year-old woman initially diagnosed with severe atopic dermatitis, who later experienced generalized itching, hyperpigmentation, alopecia, and palmoplantar keratoderma over several years. A skin biopsy revealed atypical lymphocytic infiltration with folliculotropism, confirming the diagnosis of folliculotropic mycosis fungoides. This case underscores the diagnostic challenges of this condition, which shares features with common dermatological disorders, leading to delayed diagnosis and worse prognosis. Early detection is crucial for improved treatment outcomes.
Introduction
Mycosis fungoides (MF) is the most common type of cutaneous T-cell lymphoma. It is a skin-homing T-cell lymphoma with a male-to-female ratio of 1.6–2 to 1. 1 Classically, it evolves from patches, plaques, and finally, a tumor with a protracted course lasting months to years. Many patients present with longstanding nonspecific eczema, psoriasis-form dermatitis, and other nonspecific presentations for years before a histological diagnosis of MF. These variants clinically resemble the classical form of MF. Follicular MF, pagetoid reticulosis, and granulomatous slaked skin have distinctive clinical and pathological features; therefore, they are classified as distinct variants in the World Health Organization–European Organization for Research and Treatment of Cancer (WHO-EORTIC) classification (Table 1). 2 Herein, we report a case of folliculotropic MF (FMF) in a 38-year-old woman who presented with generalized itching, melanoerythroderma, and alopecia, previously misdiagnosed as severe atopic dermatitis
WHO-EORTC classification of cutaneous lymphomas with primary cutaneous manifestations. 2

Case presentation
A 38-year-old woman presented with generalized body itching, hyperpigmentation, scalp alopecia, and lower limb edema that had persisted for 3 years. Her medical history revealed that she was relatively healthy until 7 years prior when she began experiencing a gradual onset of generalized itching, primarily on the extremities. At another hospital, a biopsy was performed and revealed eczematous changes, leading to a diagnosis of atopic dermatitis. Despite administering topical steroids and emollients for 4 years, her condition failed to improve. Over time, her pruritus worsened, and she developed significant skin darkening, prompting a diagnosis of severe recalcitrant atopic dermatitis. In an attempt to manage her symptoms, cyclosporine was prescribed for 1 year, but again, there was no improvement. In addition, she began to experience weight loss and a decreased appetite before seeking medical advice at our facility. Upon physical examination, the patient exhibited diffuse non-scarring alopecia affecting 80% of her scalp, accompanied by fine flaky scales. Her skin displayed generalized hyperpigmentation with a deep purple hue, along with fine scaling consistent with melanoerythroderma. Notably, scratches and excoriations were more pronounced on her lower limbs. The examination also revealed severe diffuse palmoplantar keratoderma with fissuring (Figure 1(a)) and bilateral pitting edema extending up to the knees (Figure 1(b)). Her nails and toes were brittle and dystrophic, characterized by yellowish discoloration due to subungual hyperkeratosis (Figure 1(c) and (d)). Several palpable lymph nodes, measuring approximately 1 × 2 cm, were noted in the cervical, axillary, and inguinal regions. No organomegaly was observed, and the remainder of the physical examination was unremarkable. The patient was subsequently admitted for further investigation. A skin biopsy revealed a dense upper and deep dermal, periadnexal, and perifollicular infiltrate consisting of atypical lymphocytes, with variable epidermotropism and folliculotropism, and occasional multinucleated giant cells were seen (Figure 2). Immunohistochemical analysis showed a profile of CD4+, CD8+, and CD7−. Peripheral flow cytometry was negative for Sézary cells, and human T-lymphotropic virus (HTLV) type I PCR was also negative. Notably, T-cell rearrangement was positive in the skin, blood, and lymph node biopsies. A general oncologist confirmed the diagnosis of FMF, classified as stage T4 N1 M0 B0. The patient was referred to a specialized oncology center for further treatment. An informed consent was taken from the patient’s husband because the patient has unfortunately passed away despite extensive therapy.
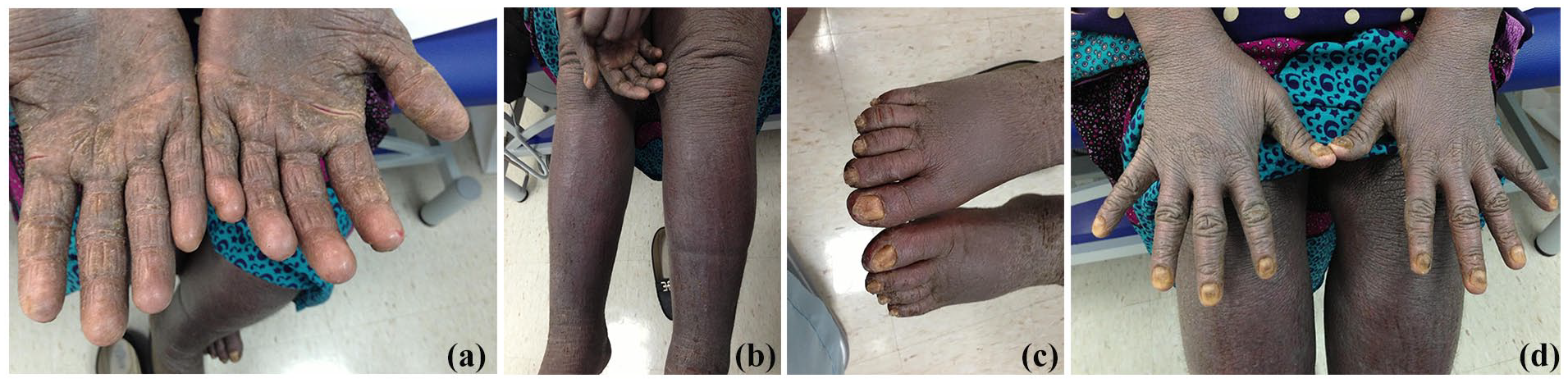
(a) Thickening of the palm with fissuresm. (b) Lower limb edema, melanoerythroderma, and excoriation. (c) Toenail dystrophy. (d) Nail changes (discoloration, dystrophy, subungual thickening), and generalized melanoerythroderma.

Skin biopsy showing follicuotrophic atypical lymphocyte.
Discussion
FMF is an aggressive variant of MF, characterized by atypical lymphocytes within the follicular epithelium and reduced infiltration of the surface epithelium. 3 It occurs more often in men, and typically affects the head and neck, particularly the scalp and face, with pruritus being a common symptom. 4 FMF can manifest as acneiform lesions, comedones, cysts, alopecia (with or without scarring), nodular prurigo-like lesions, and leonine facies. 5 These diverse manifestations often mimic other skin conditions, complicating diagnosis. Our patient initially exhibited nonspecific eczematous lesions on the lower limbs, which over 7 years developed into chronic melanoerythroderma, fissuring palmoplantar keratoderma, scalp alopecia, and dystrophic nail changes. This atypical presentation significantly delayed diagnosis, highlighting the complexities of identifying FMF, which is often confused with more common dermatologic conditions like atopic dermatitis. Key turning points in diagnosis included recognizing unusual features specifically the combination of palmoplantar keratoderma and alopecia that prompted further investigation and repeated biopsies. The implications of this delayed diagnosis are profound. According to a large cohort study, patients with FMF have a worse prognosis than classic MF and lower survival rates because they are usually at an intermediate or advanced stage at the time of diagnosis. 6 Given FMF’s poor prognosis, it requires more aggressive treatment than patients with conventional MF. The Dutch Cutaneous Lymphoma Group recommends that early-stage FMF may benefit from standard treatments used in early-stage classic MF with an excellent prognosis. Ultraviolet-B phototherapy and topical glucocorticoids are ineffective in indolent and advanced cases, while systemic treatments such as oral retinoids or Psoralen + ultraviolet light A (PUVA) photochemotherapy are more efficacious. Radiation therapy and total skin electron beam therapy are recommended for advanced indolent FMF. 7 In conclusion, FMF is an aggressive form of mycosis fungoides, often presenting with symptoms that mimic other skin conditions, leading to diagnostic delays. These delays are a significant factor in the poor prognosis associated with FMF, as patients are often diagnosed at advanced stages.
Footnotes
Acknowledgements
None.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Patient consent obtained
Unfortunately, the patient has passed away. The informed consent was then taken from the patients’ family with the understanding that photographs and medical information may be published in print and online and could be publicly available.