
Abstract
Dias-Logan syndrome, also known as intellectual developmental disorder with persistence of fetal hemoglobin (HbF), or BCL11A-related intellectual developmental disorder, is an extremely rare neurogenetic disorder characterized by intellectual disability (ID), delayed psychomotor development, variable dysmorphic features, and asymptomatic persistence of fetal hemoglobin. The prevalence and incidence of this condition are currently unknown. We report an 8-year-old Han Chinese male patient with Dias-Logan syndrome who carries a de novo heterozygous pathogenic variant, c.1078dupC (p.Leu360Profs*212), in the BCL11A gene, leading to ID and γ-globin suppression, identified through trio-based whole exome sequencing (trio-WES). All his blood parameters were normal except for an elevated HbF level, which was 19.9% of total hemoglobin. Given the negative family history for ID, epilepsy, and alcohol consumption, de novo inheritance was presumed. Consequently, trio-WES analysis (parents and child) was conducted as it can identify potential new causal variants in the offspring. So far, a comprehensive understanding of the phenotypic spectrum of Dias-Logan syndrome and the impact of genotypic variation on disease severity is still lacking. Therefore, our case report enriches the existing literature on the clinical spectrum and genotype–phenotype correlations of BCL11A-related syndrome and provides some helpful information for diagnosis, management, and genetic counseling.
Keywords
Introduction
Intellectual disability (ID) refers to a group of conditions characterized by substantial limitations in both intellectual functioning and adaptive behavior, starting before the age of 18 years. The overall incidence of ID and developmental delay is estimated to be at least 2%–3% worldwide. 1 Dias-Logan syndrome (OMIM #617101), also known as intellectual developmental disorder with persistence of fetal hemoglobin (HbF), BCL11A-related intellectual developmental disorder, or BCL11A-related syndrome, is a rare autosomal-dominant genetic defect characterized by a variety of dysmorphological features, including ID, microcephaly, delayed psychomotor development, cleft palate, strabismus, and external ear abnormalities, as well as asymptomatic persistence of HbF.2,3 Although mutations of the BCL11A gene located on 2p16.1 have been suggested to be associated with Dias-Logan syndrome,4,5 the exact prevalence and incidence of this condition remains unclear.
BCL11A, which consists of 15 exons, encodes the BCL11 transcription factor A, a regulatory C2H2-type zinc finger protein associated with the BAF SWI/SNF chromatin remodeling complex. 6 BCL11A is specifically expressed in the brain, B lymphocytes, and adult erythroid lineage, indicating its critical role in specific biological processes and functions in these tissues and cell types. BCL11A binds to the 5′-TGACCA-3′ sequence motif in the regulatory regions of target genes, including a distal promoter of the hemoglobin subunit gamma 1 gene (HBG1), and represses the switching of γ-globin to β-globin during the transition from fetal to adult erythropoiesis.6,7
Although Dias-Logan syndrome is known to be caused by heterozygous loss-of-function of variants of BCL11A,2,3 the formal clinical diagnostic criteria for this condition have not been established. Moreover, the existing understanding of its phenotypic spectrum as well as the effects of genotypic variations on disease features and severity are lacking. 3 Here, we report a case of Dias-Logan syndrome in a Han Chinese male patient with a de novo c.1078dupC (p.Leu360Profs*212) heterozygous pathogenic variant of BCL11A that caused ID and γ-globin suppression, which was identified using trio-based whole exome sequencing (trio-WES). We have also briefly summarized the BCL11A mutation spectrum related to Dias-Logan syndrome, including a total of 24 unique pathogenic variants (13 frameshift, 4 missense and 7 nonsense). We hope this case report enriches the existing literature on the clinical spectrum and genotype–phenotype correlations of BCL11A-related syndrome and provides some helpful information for the diagnosis, management, and genetic counseling of this congenital anomaly.
Case presentation
An 8-year-old Han Chinese male patient was admitted to the Department of Reproductive Medicine, Zhejiang Provincial Hospital of Integrated Traditional Chinese and Western Medicine & Hangzhou Red Cross Hospital, China for “ID and delayed language development.” The boy’s mother was G2P1, and he was born in a full-term natural delivery (birth weight, 3.30 kg; body length, 49.00 cm). He was the only child of his parents. The mother’s pregnancy and delivery history were normal. The patient had lagged behind his peers in development since childhood. He could speak and walk independently at the age of three. At presentation, he had basic living skills, fine motor disorder, severe speech difficulties manifesting as poor expression ability, and no limb shaking or convulsions. The parents denied exogenous factors such as maternal alcohol abuse during pregnancy, infections, birth complications, extreme malnutrition, and other exposure factors. The patient had no family history of ID, epilepsy, or alcohol consumption. Brain magnetic resonance imaging and chest echo Doppler imaging showed no morphological alterations. The results of metabolic tests for the patient were normal. General physical examination revealed the following findings: height, 123 cm; weight, 20.5 kg; head circumference, 50.5 cm (−0.3 SD); slightly wide nose, left auricle deformity, and asymmetric upper and lower teeth occlusion. The joints of both hands could be extended normally, and the patient showed no deformity of the spine. Wechsler Intelligence Scale for Children (WISC) testing showed the following findings: verbal comprehension index: 16, fluid reasoning index: 10, working memory index: 12, and processing speed index: 7. 8 The total WISC score was 45. The boy’s intelligence quotient was 42, indicating moderate ID. The routine blood examination showed no obvious abnormality except a high HbF level (19.9%; reference range in Chinese: <2.00%; Table 1).
Clinical signs in our patient and in the cases reported in the literature.
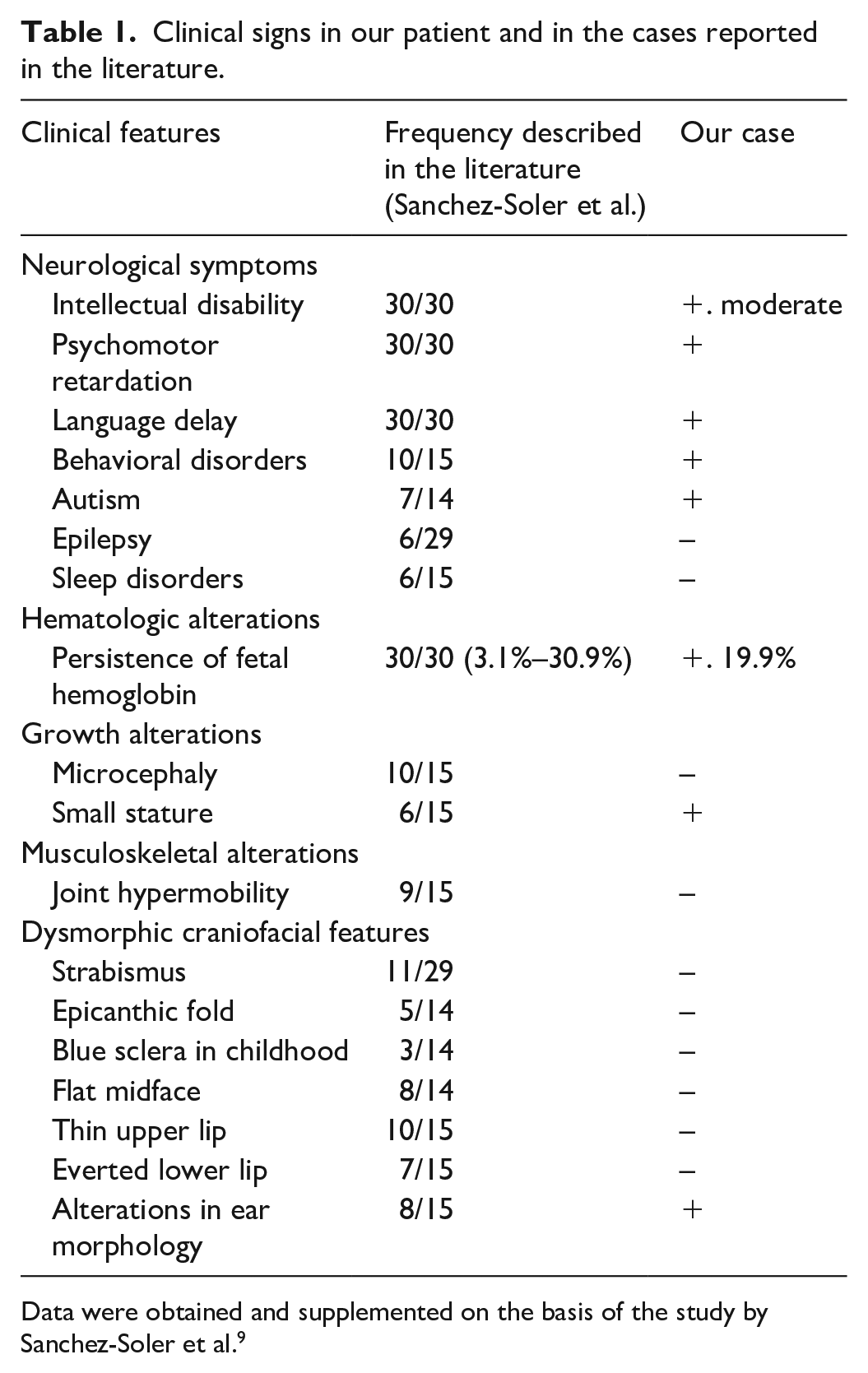
Data were obtained and supplemented on the basis of the study by Sanchez-Soler et al. 9
The parents were phenotypically and genotypically normal. Therefore, de novo inheritance was presumed, and G-banding karyotyping and array comparative genomic hybridization (array-CGH) testing, mitochondrial DNA (mtDNA) sequencing, and trio-WES were performed in the patient and the parents. The cytogenetic and cytogenomic analyses as well as mtDNA sequencing did not reveal any genetic abnormalities. Among the genetic variants of trio-WES that passed the filtering criteria of dbSNP (https://www.ncbi.nlm.nih.gov/snp/), 1000 Genomes (https://www.internationalgenome.org), gnomAD (https://gnomad.broadinstitute.org/), NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), OMIM (https://www.omim.org/), and HGMD (https://www.hgmd.cf.ac.uk/ac/index.php) databases, a de novo c.1078dupC (p.Leu360Profs*212) heterozygous pathogenic variant located in the fourth exon of the BCL11A gene appeared only in the patient and was evaluated further. Sanger sequencing verified this frameshift mutation (Figure 1). Applications that evaluated DNA sequence variants for their disease-causing potential, such as SIFT (https://sift.bii.a-star.edu.sg/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/dbsearch.shtml), and Mutation Taster (https://www.mutationtaster.org/), predicted that the variant was deleterious. On the basis of the classification of sequence variants recommended by the American Collage of Medical Genetics and Genomics, this de novo frameshift mutation in the BCL11A molecule fulfilled the criteria for a very strong and pathogenic sequence variant. On the basis of the patient’s clinical symptoms and the results of trio-WES, a diagnosis of Dias-Logan syndrome was made.

Portion of the DNA sequences of the BCL11A gene (reverse sequence) identified by Sanger sequencing. Both the father (a) and the mother (c) showed normal sequences, and only the patient (b) showed a heterozygous pathogenic variant: c.1078dupC within the fourth exon of BCL11A. The purple arrow indicates the position of the duplicated nucleotide cytosine (C).
Unfortunately, the patient was lost to follow-up immediately after diagnosis.
Discussion
Dias-Logan syndrome, which is characterized by developmental delay, ID, and persistence of HbF, is an extremely rare neurogenetic disorder that was first reported in 2016. 2 The existing literature on this disease is quite limited.2,3,10 Dias-Logan syndrome has been confirmed to be associated with autosomal-dominant mutations in BCL11A. Additionally, some affected individuals with de novo variants have been shown to exhibit epilepsy syndromes and cerebellar malformations. 11 The main clinical symptoms in our case were very similar to those reported in the literature (Table 1). The patient showed a duplicated nucleotide C mutation of BCL11A (c.1078dupC), which resulted in a premature termination codon (PTC) and a frameshift at the position 360 of leucine in the BCL11A protein: p.Leu360Profs*212.
Since translation of mRNA ceases when a termination codon is reached, a variant that converts a coding codon into a termination codon causes translation to stop prematurely. In general, mRNAs harboring a PTC are targeted for rapid degradation through a cellular process known as nonsense-mediated mRNA decay (NMD), and no translation is possible. Most of the Dias-Logan syndrome-related BCL11A variants reported are frameshift variants. In Table 2, we have briefly summarized the clinical spectrum and genotype–phenotype correlations of Dias-Logan syndrome in the literature, including a total of 24 unique pathogenic variants (13 frameshift, 4 missense, and 7 nonsense) of BCL11A, of which frameshift mutations accounted for 54.2% (Table 2).
Twenty-four Dias-Logan syndrome-related BCL11A variants in the literature.
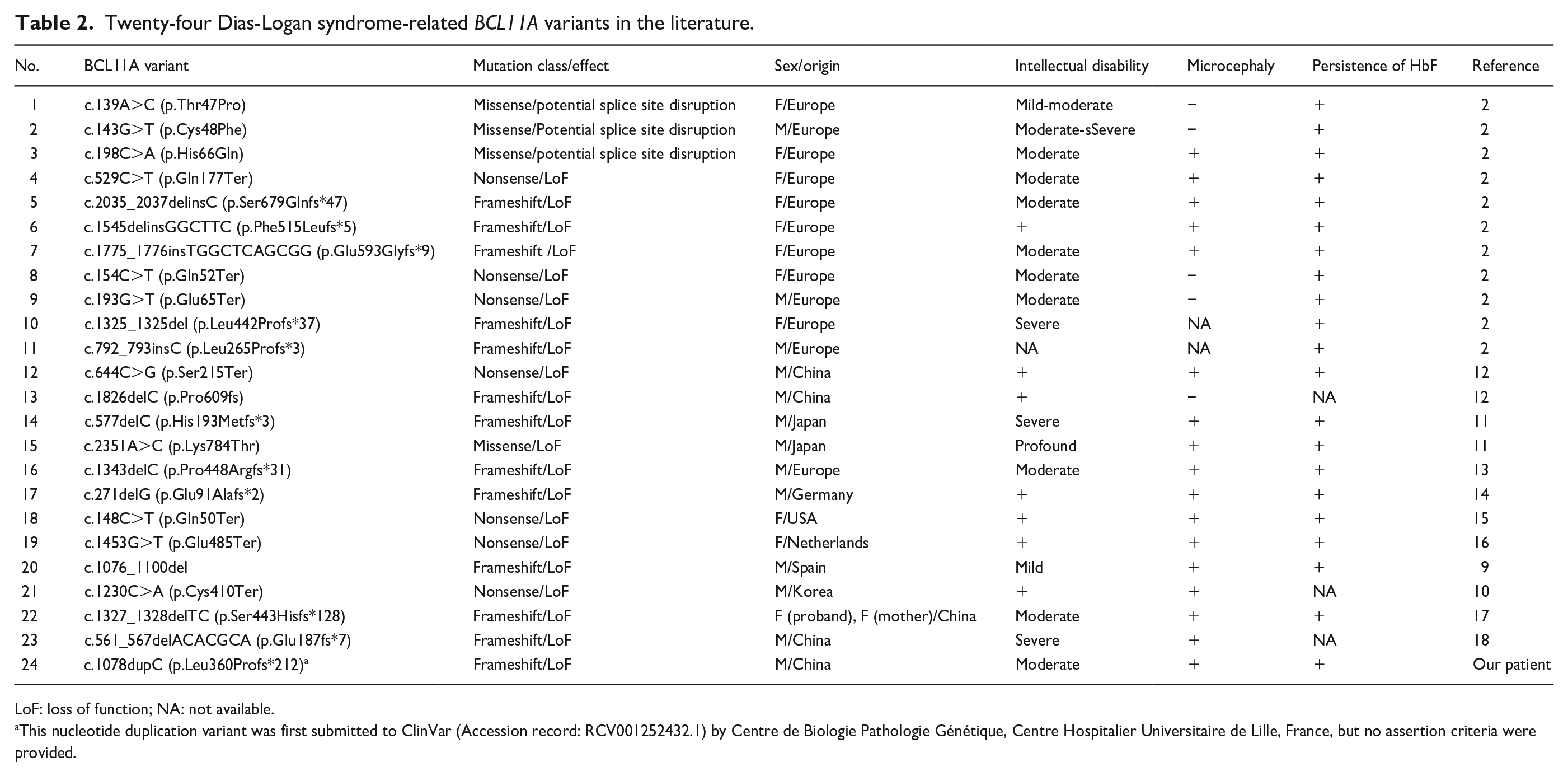
LoF: loss of function; NA: not available.
This nucleotide duplication variant was first submitted to ClinVar (Accession record: RCV001252432.1) by Centre de Biologie Pathologie Génétique, Centre Hospitalier Universitaire de Lille, France, but no assertion criteria were provided.
Peron et al. reported that the most prevalent features of Dias-Logan syndrome were ID, postnatal-onset microcephaly, hypotonia, behavioral abnormalities, autism spectrum disorder (ASD), persistence of HbF, and a previously unreported feature: autonomic dysregulation. 19 Since BCL11A is a silencer of γ-globin gene expression in adult erythroid cells, measurement of HbF levels in patients with ID, microcephaly, and epilepsy may enable earlier diagnosis, benefiting the patients and their families.1-3,19 Even for cases with a BCL11A variant of uncertain clinical significance, HbF testing by hemoglobin electrophoresis or high-performance liquid chromatography is recommended to identify Dias-Logan syndrome. 19 Among the 21 patients presenting with the 24 Dias-Logan syndrome-related BCL11A variants we analyzed, 18 showed increased HbF levels, while HbF data for the remaining three patients could not be obtained since the patients’ serum samples were unavailable.10,12,18
Experimental evidence supports the expression of three main BCL11A isoforms in the developing human brain, that is, a short BCL11A molecule consisting of 243 amino acid residues (BCL11A-S), a long 773-residue molecule (BCL11A-L), and an extra-long 835-residue protein (BCL11A-XL).2,20 Bioinformatics analysis indicated that most of the BCL11A variants may escape NMD, including c.1078dupC, which results in a significantly truncated protein. Moreover, the c.1078dupC variant affects all zinc finger clusters of only BCL11A-L and BCL11A-XL. 19 Peron et al. proposed that variants affecting all isoforms are associated with a higher frequency of hypotonia, while those affecting the BCL11A-L and BCL11A-XL isoforms and sparing BCL11A-S are associated with a higher frequency of postnatal microcephaly. 19 However, our male and a female patients in Peron’s study (P19) did not show postnatal microcephaly (Supplemental Table 1). 19
Determining the genetic cause of ID can be particularly challenging, especially in the absence of other clinical clues or information about the specific gene or region of the genome responsible. In sporadic cases without an obvious family history, a precise diagnosis can be helpful for clinical management and genetic counseling. 21 Trio-WES analysis of the parents and child is a robust method to identify new causal variants in the offspring. 22 Advanced brain imaging could also be very helpful for diagnosis and treatment planning in cases of Dias-Logan syndrome. 19
For the purposes of genetic counseling, following the diagnosis of Dias-Logan syndrome caused by an apparently de novo mutation in BCL11A, a ~4% recurrence risk quoting is recommended. 19
Symptomatic treatment is the only existing management modality available for Dias-Logan syndrome.3,19 Since our patient showed some symptoms of ASD, dextromethorphan, the only FDA-approved pharmaceutical treatment for pseudobulbar affect, may have been an option for behavioral improvement. Numerous preclinical investigations and many open-label or blinded clinical studies have demonstrated the beneficial effects of dextromethorphan across a variety of neurological and psychiatric disorders.23,24 The life span and typical causes of death in cases of Dias-Logan syndrome are unknown, with an ascertainment bias toward diagnosis in childhood. However, the lack of life-limiting congenital anomalies in affected individuals suggests a favorable long-term prognosis with appropriate support. 3
The limitations of this report include the fact that the findings are limited to a single case and that the possibility of parental germline mosaicism could not be ruled out. Future studies with a knock-in animal model of c.1078dupC variant may provide more insights into the pathogenic mechanisms.
Conclusion
Dias-Logan syndrome is a rare autosomal-dominant genetic defect without known regression of skills. In clinical practice, not all patients present with a distinctive facial phenotype, and no facial similarities are linked to specific BCL11A variants. The presence of dysmorphic features and abnormal behavior accompanying ID can help establish the diagnosis. The full range of screening methods, including HbF testing, advanced brain imaging analysis, chromosomal microarray analysis, as well as trio-WES, and whole-genome sequencing, must be considered to determine the heterozygous pathogenic variant of the BCL11A gene. The presence of a de novo variant in a child is usually additional evidence for the pathogenicity of that variant. The existing treatment modalities for this condition are primarily supportive and dictated by symptoms. In general, affected individuals without devastating congenital defects would have a supportive lifelong prognosis and outcome. Once the BCL11A pathogenic variant has been identified in an affected family member, prenatal diagnosis for a pregnancy at increased risk and preimplantation genetic testing are possible. Similar to the previously published cases, our case demonstrates the great value of clinical exome sequencing in identifying cases of nonspecific neurodevelopmental disorder with very low prevalence.
Supplemental Material
sj-xlsx-1-sco-10.1177_2050313X251314069 – Supplemental material for Dias-Logan syndrome with a de novo p.Leu360Profs*212 heterozygous pathogenic variant of BCL11A in a Chinese patient: A case report
Supplemental material, sj-xlsx-1-sco-10.1177_2050313X251314069 for Dias-Logan syndrome with a de novo p.Leu360Profs*212 heterozygous pathogenic variant of BCL11A in a Chinese patient: A case report by Yizhuo Shu, Xiaoling Chen, Zhuoqun Wei and Chunyue Chen in SAGE Open Medical Case Reports
Footnotes
Acknowledgements
The authors express their sincere gratitude to the patient and his family for their invaluable contribution to this study.
Author contributions
C.C., Z.W., and Y.S. designed the research study, analyzed the data. Y.S. drafted the initial version of the manuscript. X.C. made a substantial contribute to the data acquisition. All authors read and approved the final manuscript.
Data availability statement
All data relevant to this study are included in the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Written informed consent was obtained from the patient’s legally authorized representative (his parents) for the anonymized information to be published in this article. A copy of the written consent is available upon request from the corresponding author. All identifiable patient details were removed before publication.
Informed consent
Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.