
Abstract
Objectives:
A pair of female Malay monozygotic twins who presented with recurrent upper respiratory tract infections, hepatosplenomegaly, bronchiectasis and bicytopenia were recruited in this study. Both patients were suspected with primary immunodeficiency diseases. However, the definite diagnosis was not clear due to complex disease phenotypes. The objective of this study was to identify the causative gene mutation in these patients.
Methods:
Lymphocyte subset enumeration test and whole exome sequencing were performed.
Results:
We identified a compound heterozygous CR2 mutation (c.1916G>A and c.2012G>A) in both patients. These variants were then confirmed using Sanger sequencing.
Conclusion:
Whole exome sequencing analysis of the monozygotic twins revealed compound heterozygous missense mutations in CR2.
Keywords
Introduction
Primary immunodeficiency diseases (PID) are rare genetic diseases that result in defective immune function. 1 Common variable immunodeficiency (CVID) is a type of PID with estimated incidence of 3.8 per 100,000 live births in Denmark, 3.1 in Iceland, 2.6 in Norway, 2.17 in Taiwan, 0.7 in France and 0.6 in Spain.2,3 CVID comprises heterogeneous immune disorders such as antibody deficiency, increased susceptibility to autoimmunity and lymphoproliferation.4,5 The diagnosis and management of the disease are difficult due to the polygenic nature of CVID and similar clinical symptoms shared with the other PID.6,7 Moreover, CVID cases have been reported to inherit in both autosomal dominant and autosomal recessive patterns. 8 Owing to the wide clinical spectrum and genetic heterogeneity, genotype–phenotype relationship of CVID remains unclear. 9
Advancement of next-generation sequencing (NGS) provides a better insight into the genetic diagnosis of complex diseases.10,11 Whole exome sequencing (WES), an NGS method that targets the protein-coding regions in the genome, had been widely used in the identification of genetic mutation in rare disorders.12–14 The use of WES has enabled the identification of a single hemizygous, missense XIAP variant in X-linked lymphoproliferative disease 2 which may not be easily diagnosed using traditional genetic techniques due to unusual clinical manifestation.15,16
A pair of monozygotic (MZ) twins (P1 and P2) of non-consanguineous Malay parents (Figure 1) were recruited in this study. In the first few years of life, they had several episodes of upper respiratory tract infections, which were resolved without requiring hospital admission. The patients were first hospitalised for respiratory tract infection at the age of 7, when they presented with tachypnoea and were noted to have digital clubbing, hepatosplenomegaly, failure to thrive, bronchiectasis and bicytopenia. However, bone marrow aspiration did not suggest any malignancy infiltration. The parents and three other siblings appeared healthy (Figure 1). These patients were suspected with PID. Therefore, WES was performed in both patients to determine the causative gene responsible for the disease phenotype.

Family pedigree of the patients. Roman numerals i and ii indicate generations, squares indicate males, circles indicate females, filled circles indicate the affected MZ twins and crossed-out circles indicate the deceased. The unaffected family members were not studied. The values under each symbol represent the age in years.
Materials and methods
Subjects and ethics statement
The study protocol was approved by the Medical Research and Ethics Committee (MREC), Ministry of Health Malaysia (KKM/NIHSEC/P16-837). MZ twins suspected with PID were recruited in this study.
Lymphocyte subset enumeration
Whole blood (10 mL) from each patient was obtained in ethylenediaminetetraacetic acid (EDTA) blood tube for lymphocyte subset enumeration and DNA extraction. The patient’s whole blood (50 µL) was stained with two cocktails containing monoclonal antibodies in a BD Trucount tube, respectively: BD Multitest™ CD3 FITC (clone SK7)/CD8 PE (clone SK1)/CD45 PerCP (clone 2D1(HLe-1))/CD4 APC (clone SK3) and BD Multitest CD3 FITC (clone SK7)/CD16+ CD56 PE (clone B73.1 and clone NCA16.2)/CD45 PerCP(clone2D1(HLe-1))/CD19 APC (clone SJ25C1). The tubes were incubated for 15 min in dark. Then, the mixture was lysed with BD FACS™ Lysing solution and incubated for 10 minutes. The percentage and absolute count of the lymphocyte subsets were analysed using BD FACSDiva™ software on a BD FACS Canto™ II flow cytometer (Becton Dickinson, USA).
DNA extraction
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood using the standard Ficoll-Paque centrifugation procedure. Genomic DNA was extracted using QIAamp® DNA Mini Kit (Qiagen, Germany) following the manufacturer’s instructions. The quality of DNA samples was tested using 1% agarose gel electrophoresis. Qubit 2.0 Fluorometer (Life Technologies, USA) was used for DNA quantification.
Exome capture and sequencing
Both patients’ genomic DNA was subjected to WES, with average sequencing depth of 100× coverage, using Agilent SureSelect Human All Exon V5 (Agilent, USA) that targets 50-Mb exonic sequence.
Quality control check and processing of sequencing reads
Quality of raw sequence data from all FASTQ files was assessed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Trimmomatic was used to trim raw reads with poor base quality and sequence adapters. 17 Remaining clean reads were aligned to the human reference genome GRCh38 (https://www.ncbi.nlm.nih.gov/assembly/GCA_000001405.27) using Burrows-Wheeler Aligner (BWA). 18 SAMtools was used for sorting reads and collecting statistical metrics. 19 Next, Picard tools (http://picard.sourceforge.net/) were utilised to mark the duplicate reads and collect sequencing artefact metrics.
Variant calling and annotation
Variant calling for single nucleotide polymorphisms (SNPs) and insertions/deletions (Indels) was performed using Genome Analysis Toolkit (GATK). 20 These variants were subsequently annotated using web-based ANNOVAR (wANNOVAR). 21
Genotype concordance and discordance analysis
To determine the concordant and discordant variants in both twins, all exonic SNPs from their variant call format (VCF) files were compared. The concordant and discordant variants were extracted and annotated using BCFtools (http://samtools.github.io/bcftools) and wANNOVAR, respectively. The variants were considered concordant if the genotypes are identical in both VCF files. Whereas variants with genotypes that are unique to only one twin were categorised as discordant variants.
Variant filtering and prioritisation
Annotated variants were filtered and prioritised based on three criteria. First criterion was variant classification such as synonymous, non-synonymous, frameshift and non-frameshift mutations. Second criterion was allele frequency reported by Genome Aggregation Database (gnomAD). Third criterion was in silico protein functional prediction for variants by Sorting Intolerant From Tolerant (SIFT), 22 Polymorphism Phenotyping v2 (PolyPhen-2) 23 and MutationTaster. 24
Polymerase chain reaction and Sanger sequencing
The CR2 variant identified by WES was confirmed by Sanger sequencing. The results were viewed using CLC Genomics Workbench v7.5.1 software.
Results
Immunological analysis
The initial diagnostic assessment revealed reduced T, B and natural killer (NK) cell counts in both patients (Figure 2 and Table 1). We found that the total T-cell values were low, 671 × 106/L and 545 × 106/L for P1 and P2, respectively. In addition, the total B-cell values were also low, 26 × 106/L and 16 × 106/L for both patients.
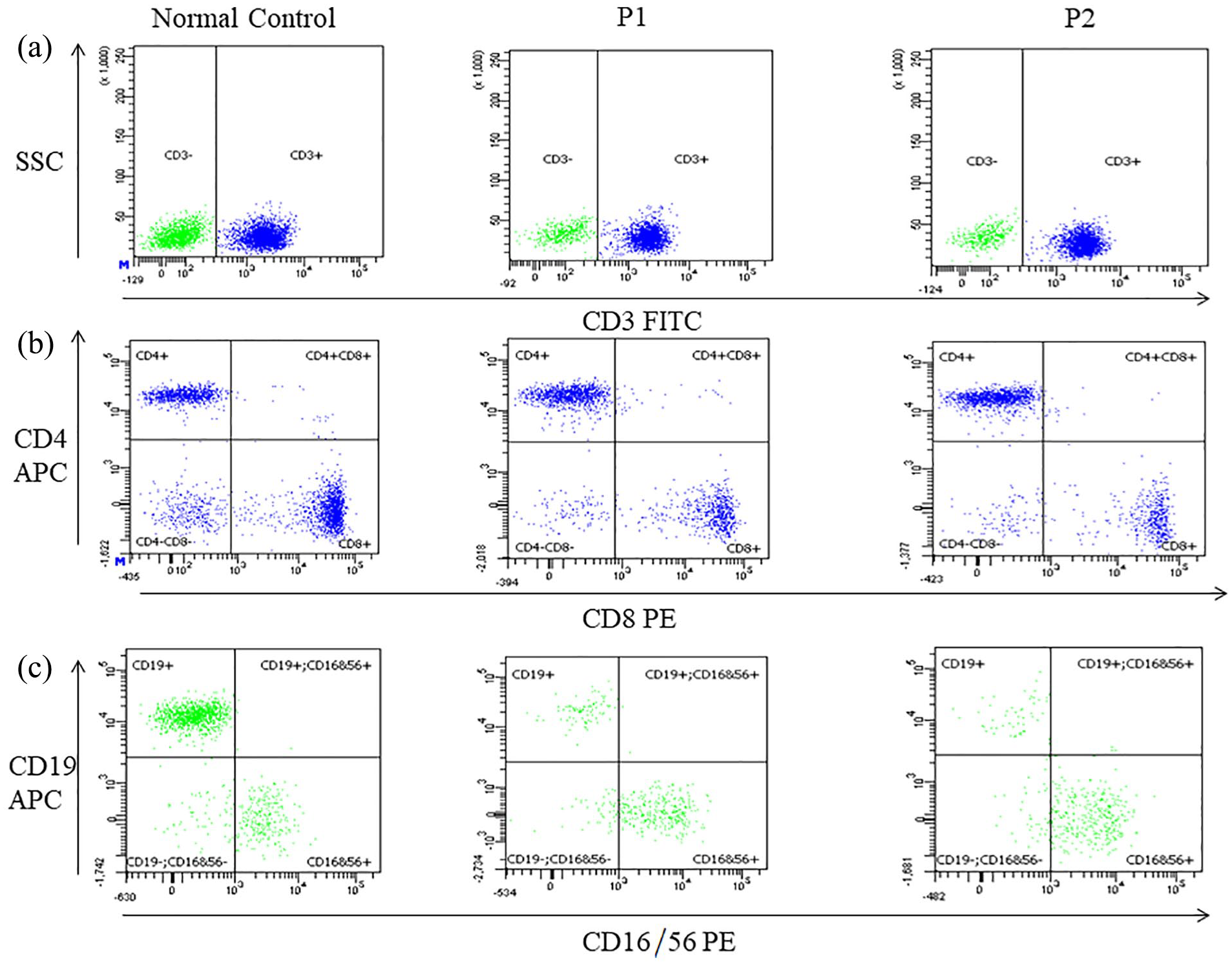
Flow cytometry data on lymphocyte subset enumeration. Dot plots show the distribution of lymphocyte sub-populations in the MZ twins and a normal control in (a) CD3+ T cells versus SSC; (b) CD3+ CD8+ T cells versus CD3+ CD4+ T cells; and (c) CD16+ CD56+ NK cells versus CD19+ B cells. Each dot represents an individual cell analysed by the flow cytometer.
Clinical and laboratory findings of the patients.

IVIG: intravenous immunoglobulin therapy; URTI: Upper respiratory tract infection; N: age-matched reference range; NK: natural killer.
WES analysis
WES generated 78,835,626 and 77,270,546 paired-end reads in P1 and P2, respectively. The reads were mapped to the human reference genome GRCh38. Both patients had achieved high percentage of mapping, which were 99.26% for P1 and 99.39% for P2. We investigated variations such as SNPs and Indels in both patients as these are the two largest types of human genetic variations. 25
Evaluation of genotype concordance and discordance
We identified approximately 22,000 exonic SNPs in both twins, 93.4% and 90.5% of which were concordant between the twins, respectively (Figure 3(a)). Besides, we also investigated the number of private and shared SNPs between each twin and an unrelated control. Conversely, when compared with an unrelated control, only 59%–64% of exonic SNPs were found concordant (Figure 3(b) and (c)).

Genotype concordance and discordance of two samples. Venn diagrams depict the number of shared and private SNPs identified between (a) P1 and P2, (b) P1 and unrelated control and (c) P2 and unrelated control.
PID gene screening
We screened the WES datasets for 240 PID-related genes as reported by the International Union of Immunological Societies for Primary Immunodeficiency to identify the genetic variations in PID-related genes. 26 We interpreted the candidate variants according to the standards and guidelines as recommended by American College of Medical Genetics and Genomics and the Association for Molecular Pathology. 27 Since the patients’ clinical symptoms resembled CVID, we narrowed the analysis to the 31 genes known to be associated with CVID. We identified 17 missense SNPs (Table 2) and 15 silent SNPs in CVID-associated genes from the WES datasets of both patients. The deleterious effect of missense SNPs on resulting proteins was evaluated using SIFT, PolyPhen-2 and MutationTaster. Homozygous SNPs were identified in CD19, CD27, CD86, ADAM28 and STXBP2. However, the damaging variant prediction tools predicted them as non-damaging variants (Table 2). Three missense SNPs (two SNPs in CR2 and one SNP in FANCA) were predicted to be damaging variants by at least one of the variant prediction softwares (Table 2). A heterozygous SNP in FANCA (rs1800282) identified in both patients was not considered as disease-associated variant because FANCA has been associated with autosomal recessive inheritance. 28 Two SNPs in CR2, c.1916G>A (rs17615) and c.2012G>A (rs17616), were identified in both patients (Table 2). Subsequent Sanger sequencing revealed that the patients’ father carried c.2012G>A (rs17616) SNP, while the mother carried c.1916G>A (rs17615) SNP (Figure 4). This indicated that both patients inherited the alleles from each of their parents leading to compound heterozygosity status.
Summary of missense variants found in the CVID-related genes.

Chr: chromosome; AA: amino acid; NP: non-polar; Pr: polar; Hom: homozygous; Het: heterozygous; T: tolerated; D: damaging; P: polymorphism; N: neutral; MAF: minor allele frequency; gnomAD: Genome Aggregation Database; B: benign.

Confirmation of compound heterozygous CR2 variants identified from WES using Sanger sequencing. DNA sequence chromatograms illustrate the variants in CR2 at (a) c.1916G>A and (b) c.2012G>A for normal control, P1, P2, mother and father of the MZ twins. The patients’ mother has a heterozygous c.1916G>A variant whereas the father has a heterozygous c.2012G>A variant, leading to the compound heterozygosity in patients.
Discussion
Here, we performed WES analysis on a pair of MZ twins suspected with PID to identify the causative genetic variation responsible for their condition. WES results demonstrated that CR2 gene could be the possible disease-associated gene in both patients. CR2 deficiency is known to be a causative factor for CVID phenotype. 29 CVID is a primary immunodeficiency characterised by a decrease in serum IgG level, a decrease in either IgA or IgM, and a poor response to vaccines in children of at least 2 years of age, after excluding other causes of hypogammaglobulinemia. 30
Complement receptor 2 (CR2), an essential receptor for Epstein–Barr virus, that expressed primarily on B cells and follicular dendritic cells was also reported on T cells. 31 This 145-kDa membrane protein is encoded by CR2 gene with 19 exons at chromosomal location of 1q32. 32 CR2 protein functions as a coordinator for humoral response and complement system. 33 The CR2 protein is composed of 15 or 16 extracellular short consensus repeats (SCRs), one transmembrane domain and an intracytoplasmic region. 34 Various types of mutations (nonsense, frameshift, splice site and missense) have been reported across all domains of CR2 protein. 32 The mutation sites (c.1916G>A and c.2012G>A) found in our patients occurred in the exon 10 and exon 11 of CR2 gene. These mutations were predicted to cause amino acid changes in the protein-coding sequence (p. Ser639Asn and p. Arg671His), respectively, in which the resulting amino acid residues are located in the conserved sushi domain or SCRs of CR2 protein. However, protein expression analysis was unable to be conducted because both patients deceased in the middle of the study. Similarly, functional assays (in vitro or in vivo) which are crucial to evaluate the pathogenicity of causative gene especially in compound heterozygous case were unable to be performed.
CR2 deficiency has been reported as autosomal recessive disorder. 26 Besides, a compound heterozygous mutation in CR2 gene was recently described to be associated with CR2 deficiency. Hypogammaglobulinemia and reduced memory B cells were noted in the patient with CR2 deficiency. 32 In our study, Sanger sequencing showed that each of our patients inherited a mutated allele from each parent, suggesting compound heterozygous status in both patients. Therefore, these variants (c.1916G>A and c.2012G>A) are most likely to be associated with the disease phenotypes seen in our patients. In addition, copy number variation (CNV) analysis in patients with complex phenotypes could be used to further investigate the concordant and discordant CNVs between MZ twins.
Conclusion
In summary, this report describes an analysis based on WES dataset in a pair of Malay-descendant MZ twins. Based on evidence from Sanger sequencing and bioinformatics analysis, the compound heterozygous missense mutations (exon 10: c.1916G>A and exon 11: c.2012G>A) in CR2 gene resulted in a change to unrelated amino acids (p. Ser639Asn and p. Arg671His) that may cause CR2 deficiency in both patients.
Footnotes
Acknowledgements
The authors would like to thank the Director General of Health Malaysia for his permission to publish this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
Ethical approval for this study was obtained from the Medical Research and Ethics Committee (MREC), Malaysia (Ethic approval no: KMM/NIHSEC/P16-837).
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research work was financially supported by the Ministry of Health, Malaysia research grant (NMRR-16-892-31023).
Informed consent
Written informed consent was obtained from all subjects before the study