
Abstract
ATRX gene (alpha-thalassemia mental retardation X-linked) encodes for a chromatin remodeler and regular transcription protein, part of the SNF2 family of chromatin remodeling proteins. Mutations in this gene have been associated with severe syndromes, including intellectual disability, typical facial dysmorphia, urogenital anomalies, and atypical alpha thalassemia. In this report, we present a 7-year-old Moroccan boy with severe intellectual disability, autistic features, typical facial dysmorphia, bilateral cryptorchidism, and scoliosis. Whole exome sequencing identified a missense variant of uncertain significance in the ATRX gene (NM_000489.3: c.745G>A). In silico tools strongly predict the pathogenicity of this variant. Moreover, this variant occurs in a highly conserved domain, potentially affecting the function of the encoded protein, and the glycine at position 249 is well conserved across different species. Further studies are needed to confirm the pathogenicity of this novel variant to establish adequate genetic counseling.
Introduction
ATRX gene (alpha-thalassemia mental retardation X-linked), discovered in 1995, 1 is a chromatin remodeler and regular transcription gene that belongs to the SNF2 family of chromatin remodeling proteins. To date, more than 150 ATRX mutations have been described in the literature. Missense mutations are the most frequently reported. 2 The alpha-thalassemia X-linked intellectual disability syndrome (ATR-X syndrome) exhibits mutations in the ATRX gene, a hereditary X-linked disease that associates severe intellectual disability, typical facial dysmorphia, microcephaly, urogenital anomalies, and atypical alpha-thalassemia.3,4
ATRX protein contains two highly conserved domains: a plant homeodomain (PHD) zinc finger at the N-terminus and a C-terminal ATPase/helicase domain. Mutations in either of these domains can lead to ATR-X syndrome. The PHD finger domain shares homology with the PHD domains of Dnmt3a, Dnmt3b, and Dnmt3L, which we refer to as the ATRX-Dnmt3-Dnmt3L (ADD) domain. Almost half of the patients with ATR-X syndrome present mutations in this domain. 5
Herein, we report a novel variant of uncertain significance in the ATRX gene in a patient with severe intellectual disability, autistic features, facial dysmorphia, and urogenital anomalies.
Case report
The patient is a 7-year-old Moroccan boy, the second of three siblings, born from a non-consanguineous marriage. The boy was born at term with a general neonatal hypotonia. He presents severe intellectual disability with autistic features, psychomotor delay, and language impairment. Clinical examination revealed a short stature (−1 SD), hypotonic face, facial dysmorphia (brachycephaly, short sling, horizontal eyebrows, bilateral epicanthus, flat nasal bridge, small triangular nose, broad columella, anteverted nostrils, cupid-bow shaped upper lip, helix badly hemmed) (Figure 1), bilateral single transverse palmar creases, bilateral cryptorchidism, and scoliosis.
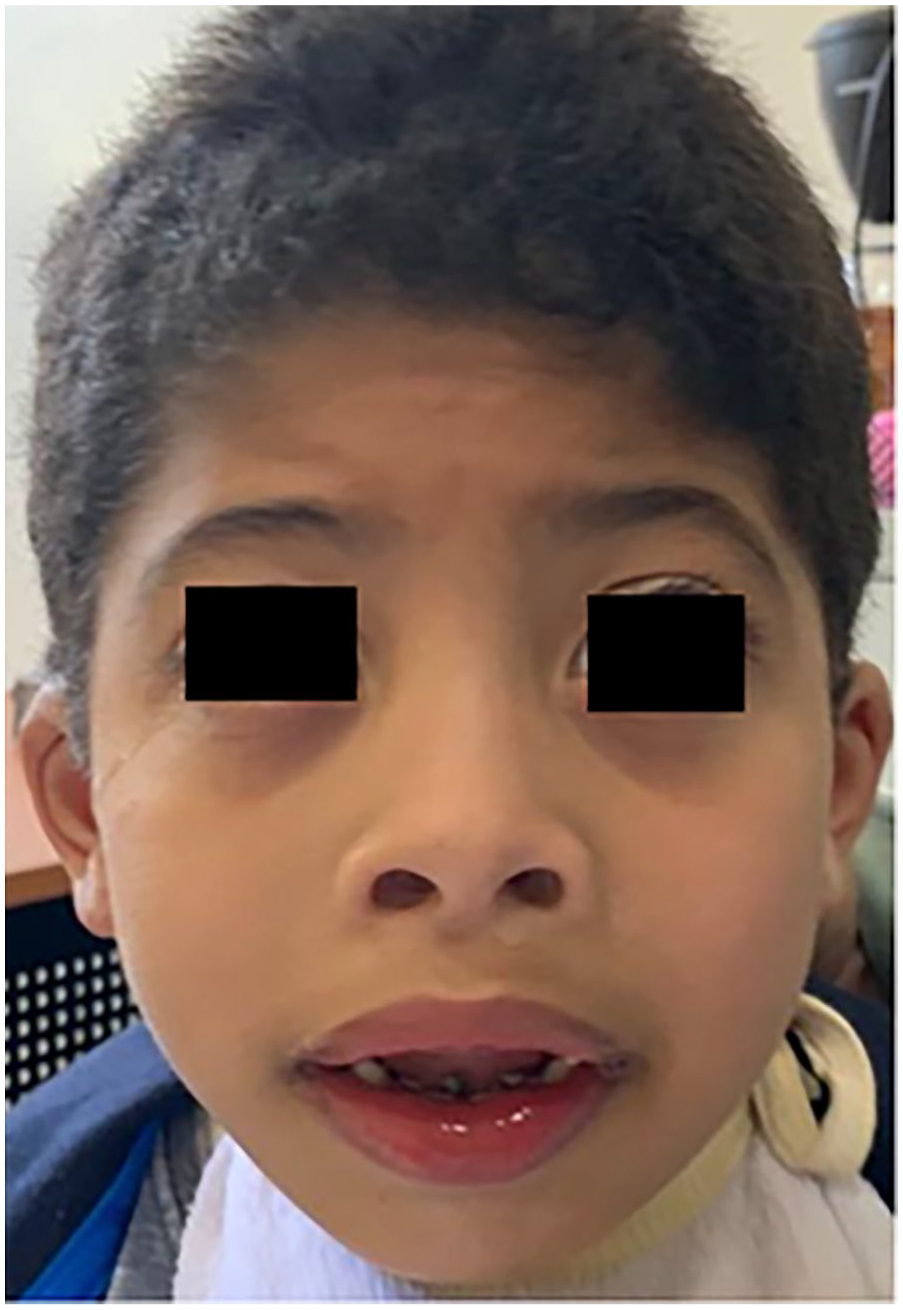
Frontal view of the patient.
Informed consent was obtained from the patient’s legally authorized representative for the use of his photo and the publication of this case report. This manuscript complies with the applicable CARE (CAse REports) guidelines.
No specific abnormalities have been identified in biology reports, electroencephalogram, auditory evoked potential, echocardiography, and electrocardiogram. Computed tomography scan and magnetic resonance imaging of the brain have shown only cortical atrophy. Constitutional karyotype and Comparative Genomic Hybridazation (CGH) array (CentoArryCyto™-750K, Centogene, Germany) were without abnormalities.
Whole exome sequencing (WES) analysis was performed. The human coding exome was sequenced targeting 98% of the coding RefSeq from the human genome build GRCh37/hg19 and completed by a paired-end sequencing with at least 20× coverage depth for 98% of the targeted areas. The results revealed a hemizygous missense variant in the ATRX gene (NM_000489.3: c.745G>A (p.(Gly249Ser)). The automated interpretation of the variant according to American College of Medical Genetics and Genomic (ACMG) clinical interpretation is uncertain signification (class 3).
In silico tools strongly predict the pathogenicity of this variant (Table 1): polyphen2 (http://genetics.bwh.harvard.edu/pph2/), SIFT program (Sorting Intolerant From Tolerant; https://sift.bii.a-star.edu.sg/), and mutation taster (https://www.mutationtaster.org/). Moreover, the alignment of the sequence containing Glycine residue at position 249 was performed across 12 species using UniProt (https://uniprot.org/Align). The protein alignment showed a high conservation for the mutated amino acid in the ATRX protein among different species (Figure 2).
In silico analysis results.


Evolutionary conservation of the glycine amino acid at position 249 of ATRX gene across 12 species.
To the best of our knowledge, this variant has never been reported in ExAC (Exome Aggregation Consortium), gnomAD (Genome Aggregation Database), or clinivar databases.
On the other side, another case has been reported to exhibit a missense variant of uncertain significance at the same position but substituting glycine for cysteine (Gly249Cys (PMID: 10204841)). The patient presents the same clinical features as our case 6 (Table 2).
The phenotype of the present patient compared with a previously reported patient.

At the protein level [uniPro: P46100], the substitution is located in a functional domain, the ADD domain, which plays a role in the establishment and/or maintenance of a normal DNA methylation pattern.
Regrettably, genetic testing for the variants in the parents was not possible because of resource limitations. However, patient follow-up has been initiated including speech therapist, rehabilitation, child psychiatrist, pediatrician, etc.
Discussion
WES analysis has proven to be an important tool for heterogeneous genetic disorders such as intellectual disability. Several de novo variants and exome-wide significant genes have been elucidated, contributing to a better comprehension of the genetic origins of intellectual disability. Moreover, de novo variants are generally considered more deleterious than inherited variants because they have been subjected to less stringent evolutionary selection. 6
The ATRX gene, located on the mammalian X-chromosome, 7 contains 37 exons that encode a 2492 amino acid protein. ATRX protein is a chromatin remodeler and regular transcription protein that belongs to the switch/sucrose non-fermentable (SWI/SNF) family. It facilitates DNA replication in multiple cellular environments and is required for efficient replication of a subset of genomic loci. 4 Null mutations of ATRX in mice result in embryonic lethality due to defects in extraembryonic trophoblast formation. In fact, disruption of chromatin remodeling is responsible for the development of cancer processes. ATRX is a typical example of a gene mutation that causes cognitive disability and cancer in sequence. 4
ATRX protein contains three primary domains: (1) the N-terminal, ADD domain (2), a centrally-located SNF2-related domain, and (3) the C-terminal SNF2 helicase-like, ATPase domain (ATRXATP). 4
Our patient presents a missense variant of uncertain significance located in exon 9 that causes the substitution of guanine for adenine at position 745 resulting in the substitution of glycerin for serine at position 249 at the protein level. This variant is localized in the middle of the ADD domain (ATRXADD) [Interpro: P46100]. 8 Pathogenic variants that affect the ATRXADD produce severe psychomotor impairment and urogenital anomalies. 9
The ATRXADD is a hybrid domain that includes a GATA-like zinc finger and a PHD finger. This unique structural motif is found exclusively in ATRX and DNA methyltransferases (DNMT3A, DNMT3B, and DNMT3L). ATRXADD is a methyl-lysine binding domain that exhibits high affinity for histone 3 when lysine 9 is trimethylated and lysine 4 is unmodified (H3K9me3/K4me0). 5
Moreover, ATRX is an X-linked syndrome predominantly affecting males. It includes several features such as severe intellectual disability, facial dysmorphism, decreased expression of the α-globin genes (thalassemia), urogenital dysfunction, and skeletal abnormalities.9,10 On the other side, female carriers with heterozygous mutations inactivated perfectly the mutated X chromosome, without any features. It has been identified in the same female with a heterozygous mutation with the X-linked disorder of ATRX syndrome. 3 No racial or ethnic concentration of individuals has been reported.
Conclusion
The routine uses of WES demonstrated some limitations, especially in some situations like intellectual disability. The challenge is the interpretation of the observed variant to confirm the diagnosis. Here, we report a variant of uncertain significance in the ATRX gene (c.745G>A (p.Gly249Ser)). In silico analysis results and the critical localization of the variant support its pathogenicity. However, further studies are needed to confirm this finding and to establish adequate genetic counseling.
Footnotes
Acknowledgements
The authors gratefully acknowledge the patient and his parents for their cooperation.
Author contributions
B.S., D.H., and T.A. conceived of the presented idea. B.S. and T.A. collected the data and drafted the manuscript. G.B. reviewed the manuscript. D.H. supervised this work. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Written informed consent was obtained from the legally authorized representative of the patient for his anonymized information to be published in this article. This case report contains no identifiable information about the patient. Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.