
Abstract
Childhood linear immunoglobulin A bullous dermatosis is a well-recognized autoimmune blistering dermatosis that can be idiopathic, drug-induced, secondary to autoimmune diseases, malignancies, infections, or gastrointestinal diseases including inflammatory bowel disease. However, it has not been previously associated with a history of organ transplantation. Here, we report two cases of childhood linear immunoglobulin A bullous dermatosis in young infants following multivisceral organ transplant, including one with a particularly recalcitrant course. We propose potential mechanisms contributing to the development of linear immunoglobulin A bullous dermatosis and challenges in the management of these patients in light of their complex medical histories and immunosuppression.
Keywords
Introduction
Linear immunoglobulin A bullous dermatosis (LABD) is a rare autoimmune blistering disorder characterized by immunoglobulin A (IgA) deposition along the basement membrane zone (BMZ). 1 Clinically, LABD presents with tense blisters and erosions, often in a rosette or annular pattern, affecting both adults and children. 1 It is associated with autoimmunity, malignancies, and gastrointestinal diseases due to IgA mucosal immune system activation. The link between LABD and transplantation is poorly understood, with few reported cases. Here, we present two unique cases of childhood LABD following transplantation.
Case 1
A 16-month-old female with a history of megacystis microcolon intestinal hypoperistalsis syndrome underwent an orthotopic multivisceral transplant involving the liver, small intestine, pancreas, and stomach in 2021. Post-transplant, she developed progressive eosinophilia while on trimethoprim-sulfamethoxazole (TMP-SMX) for pneumocystis pneumonia prophylaxis, leading to a switch to dapsone 2 mg/kg and amoxicillin for asplenia. Her immunosuppression regimen included mycophenolate mofetil, tacrolimus, sirolimus, and prednisone 0.1 mg/kg.
Three months post-transplant, she developed a widespread erythematous and bullous eruption, particularly around her Broviac site, accompanied by a fever of 38.3°C. She was empirically treated with piperacillin-tazobactam and vancomycin, but cultures returned negative. Initially improving on vancomycin, her condition worsened after switching to cefazolin, developing more vesicles and pruritic erythematous plaques, but were responsive to antihistamines. A skin biopsy revealed a subepidermal blister with neutrophils and strong linear IgA positivity at the BMZ, consistent with LABD. The bullous autoimmune profile also returned positive for desmoglein 3.
A medication review identified amlodipine as a potential drug trigger. As part of her initial management, prednisone was increased to 1 mg/kg/day, and amlodipine was replaced with enalapril. While on dapsone (2 mg/kg), she developed desaturations and methemoglobinemia, requiring methylene blue treatment and pulse methylprednisolone 1 mg/kg/day. Dapsone was restarted at 1 mg/kg/day and was gradually increased to 1.3 mg/kg/day.
Despite an initial course of intravenous immunoglobulins (IVIG) 1 g/kg, she continued to have ongoing pruritus and blistering, which was managed symptomatically with oral rupatadine, and eventually was switched to oral hydroxyzine, and oral doxepin. Her topical and wound care regimens were adjusted to include Protopic 0.03% ointment, Betaderm 0.05% ointment for pruritic lesions, Clobetasol 0.05% ointment for prodromal blister, mupirocin 2% ointment for open sores, and covered with Adaptic, gauze, and Kling or BurnNet. Flovent was sprayed on the open lesions to improve bandage adhesion.
While admitted, she developed several infection-related LABD flares. She required a second IVIG course (1 g/kg) and an increased dapsone dose (2 mg/kg/day) during a Norovirus infection. The blistering persisted until she received 4 doses of rituximab, which controlled the disease and allowed for tapering of prednisone and dapsone. She had another flare with a urinary tract infection (UTI) but a request for additional rituximab was denied, so she was treated symptomatically with meropenem and topical steroids which was switched to topical dapsone 5% gel post-UTI. Due to residual severe pruritus, she received omalizumab but developed emesis after the first infusion but resolved within 24 h without recurrence.
She had another flare due to an intestinal prolapse, requiring surgical stoma reduction. She received a third dose of IVIG and a fifth dose of rituximab, with aspirin being discontinued due to being a potential LABD trigger.
Post-discharge, the patient had recurring but less severe flares, managed with prednisone. Acetaminophen, given PRN for every illness or fever, was identified as a potential trigger. Reducing acetaminophen usage led to significant improvement, allowing discontinuation of prednisone and tapering of dapsone. She now uses antihistamines as needed for pruritus.
Case 2
An 11-month-old female with biliary atresia and cirrhosis underwent a multivisceral transplant, including the small intestine, pancreas, and liver in 2022. Her immunosuppressive regimen included tacrolimus, mycophenolate mofetil, and prednisone 0.5 mg/kg/day, following the cessation of sirolimus and TMP-SMX due to suspected drug-induced IgA nephropathy. About 3 months post-transplant, she developed a blistering eruption (Figure 1(c) and (d)), unresponsive to increased prednisone (1 mg/kg/day). A skin biopsy revealed subepidermal bulla with neutrophils and 2+ linear IgA positivity, consistent with LABD. Treatment included dapsone at 0.8 mg/kg, Betaderm 0.1% ointment, and wound care.

(a) Extensive erythematous and bullous eruptions around the ostomy, (b) Broviac site in Case 1, and (c, d) similar eruptions on the leg and torso in Case 2.
Despite improvement, a new vesicle appeared 4 weeks later coinciding with the reintroduction of sirolimus and enalapril. Both medications were temporarily held but were resumed with no further issues, with these new vesicles being attributed to Koebnerization. Prednisone was increased to 2 mg/kg/day by nephrology for IgA nephropathy, and dapsone was maintained at 1 mg/kg/day without complications. She was eventually discharged with topical dapsone 5% gel twice daily and tapering of oral dapsone and prednisone as an outpatient with no further issues.
Discussion
In our cases, both patients presented with clinical and histological findings consistent with LABD, manifesting 3 months after multivisceral transplantation. Both were female, aligning with LABD epidemiology, but markedly younger than the reported mean age of 4.5 years. Patient 1 had a complex course, with multiple infections exacerbating LABD. Patient 2 had a more predictable treatment course, responding well to dapsone, though Koebnerization—rare in LABD—was observed.
Due to the refractory nature of case 1, other treatments were considered based on the literature review. Tocilizumab was considered but required confirmation of IL-6 levels in blister fluid. 2 Cloxacillin was also considered but not used due to limited evidence. Acetaminophen emerged as a potential trigger, though not widely recognized, with few cases reported. 3
Our literature search revealed eight cases of transplant-related LABD—one in a child and seven in adults—occurring between 72 days and 11 years post-transplant (Table 1). Two cases were in hematopoietic stem cell transplant recipients for leukemia and lymphoma, suggesting a paraneoplastic origin rather than a direct transplant relation.4,5 Another case was linked to vancomycin, a known LABD trigger. 6 The remaining cases involved solid organ transplants and were mostly idiopathic.3,7–10
Case reports of transplant-associated linear IgA bullous dermatosis.
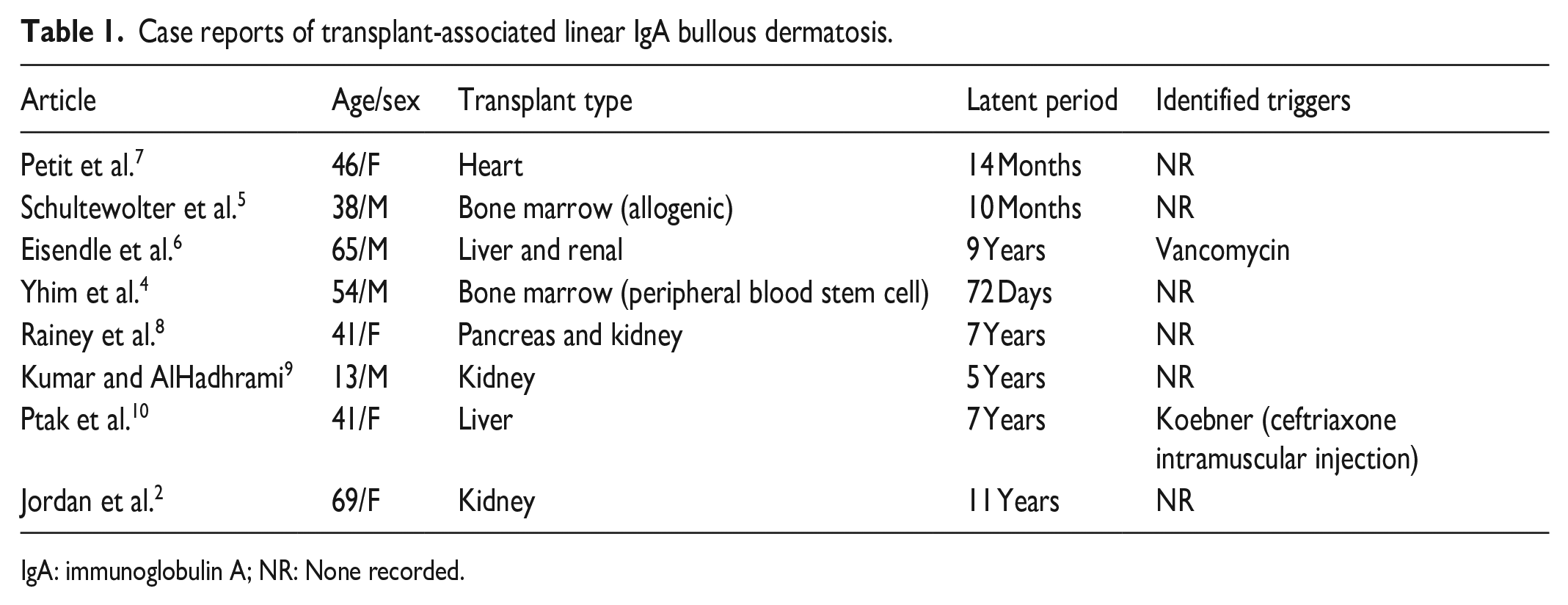
IgA: immunoglobulin A; NR: None recorded.
Several mechanisms might explain LABD in transplant patients, with immune dysregulation playing a key role. One potential mechanism is graft-versus-host disease (GvHD), where donor cytotoxic T cells could expose epidermal BMZ antigens, leading to IgA synthesis and autoantibody formation. 11 The 90-day latency period for LABD observed in our cases aligns with the timeline for chronic GvHD and humoral immune response development. Additionally, the only coinciding factor in both patients at the 3-month mark when LABD occurred was a routine reduction in post-transplant immunosuppression further suggesting the mechanism of immune dysregulation.
In summary, these cases highlight a possible association between LABD and solid organ transplantation, a rare phenomenon with unclear causation. Further research is needed to understand its mechanisms, risk factors, and optimal prevention and management strategies.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Consent for publication
Consent for the publication of non-identifying patient photographs and material was obtained by the authors and included at the time of article submission to the journal stating that all patients gave consent with the understanding that this information may be publicly available.