
Abstract
Hereditary angioedema (HAE) is a rare autosomal dominant condition characterized by C1-INH gene mutations, leading to recurrent angioedema episodes affecting various body parts, including the gastrointestinal tract. This case report describes a 24-year-old female presenting with symptoms mimicking an acute abdomen, characterized by severe abdominal cramps, anorexia, and diarrhea, with a significant past medical history of angioedema flares and emergency intubation for asphyxiation at age 11. Despite initial treatment with antihistamines showing no improvement, her symptoms spontaneously resolved. Further investigation revealed low complement C4 levels and reduced C1-INH function, confirming HAE with an unusual isolated involvement of the ascending and transverse colon. This case underscores the importance of considering HAE in patients presenting with acute abdominal symptoms, especially with a history suggestive of angioedema. It highlights the need for emergency physicians and gastroenterologists to be aware of HAE’s clinical manifestations to avoid misdiagnosis and unnecessary interventions. Moreover, the case emphasizes the significance of patient education on recognizing symptoms and seeking timely medical attention to prevent severe complications. This report adds to the existing literature by detailing an uncommon presentation of HAE, aiming to enhance early diagnosis and management of this potentially life-threatening condition.
Introduction
Angioedema refers to localized, non-pitting swelling of the subcutaneous and/or submucosal tissues, impacting areas such as the lips, face, neck, limbs, oral cavity, larynx, and gastrointestinal tract, and can be classified as an acquired or hereditary form. 1 Hereditary angioedema (HAE) is a rare autosomal dominant disorder that results from mutations in the C1-INH gene on chromosome 11.2,3 The incidence of HAE falls between 1:10,000 and 1:150,000. 2 There is not yet any information on differences in HAE prevalence by gender or ethnicity. 4 HAE is characterized by repeated episodes of angioedema, impacting the skin, gastrointestinal tract, or upper respiratory tract. 2
When HAE affects the upper respiratory tract, it can be fatal due to the risk of asphyxiation, while its non-specific gastrointestinal symptoms could lead to misdiagnosis and unnecessary procedures, highlighting the importance of accurate diagnosis. 5
This case report provides valuable insights into a rare presentation of HAE, where colon swelling mimicked acute abdomen symptoms in a 24-year-old female. It underscores the diverse clinical spectrum of HAE and highlights the complexities in diagnosis, emphasizing the need for comprehensive evaluation and interdisciplinary collaboration. By sharing this experience, we aim to enhance awareness and promote early recognition of rare conditions like HAE, ultimately improving patient outcomes.
Case presentation
A 24-year-old woman presented to the Emergency department complained of severe abdominal cramps and diarrhea for 2 days, the patient denied any combined symptoms of skin rash, swelling over the face, generalized urticaria, difficulty breathing, nausea, or vomiting. She denied any drug use recently, travel history, or sick contacts. She had one episode of fever 3 days before the presentation. The maximum degree was 39°C, and she had a loss of appetite for 1 week. Her medical history was significant for angioedema flares when she was young, her face, larynx, and tongue were affected and treated with danazol, an androgen derivative discontinued 5 years ago due to adverse effects of breathlessness, weakness, and amenorrhea. At 11, she underwent emergency intubation for asphyxiation. Her medical family history was not significant for similar presentation or a hereditary medical condition. Upon examination, her vitals were stable and afebrile, but her abdomen was distended, and tender, with hyperactive bowel sounds, with no skin rash, urticarial, or swelling of face and tongue noted. Laboratory results showed a WBC count of 6.2 × 109/L, C-reactive protein of 4 g/L, serum ferritin 102 ng/mL, stool culture was negative for both Salmonella and Shigella, and urinalysis was negative. She was admitted with a suspected acute abdomen, she was kept nil per oral, given intravenous fluids for hydration, Perfalgan (paracetamol) for pain control, Pantover (Pantoprazole), and intravenous antibiotic ceftriaxone was initiated empirically. Abdominal computed tomography (CT) findings were conducted. A significant wall thickening with submucosal edema affecting the ascending colon was found (Figure 1), in addition to the affected proximal transverse colon with an abrupt transformation into a normal colon (Figure 2). Endoscopy showed no significant findings.
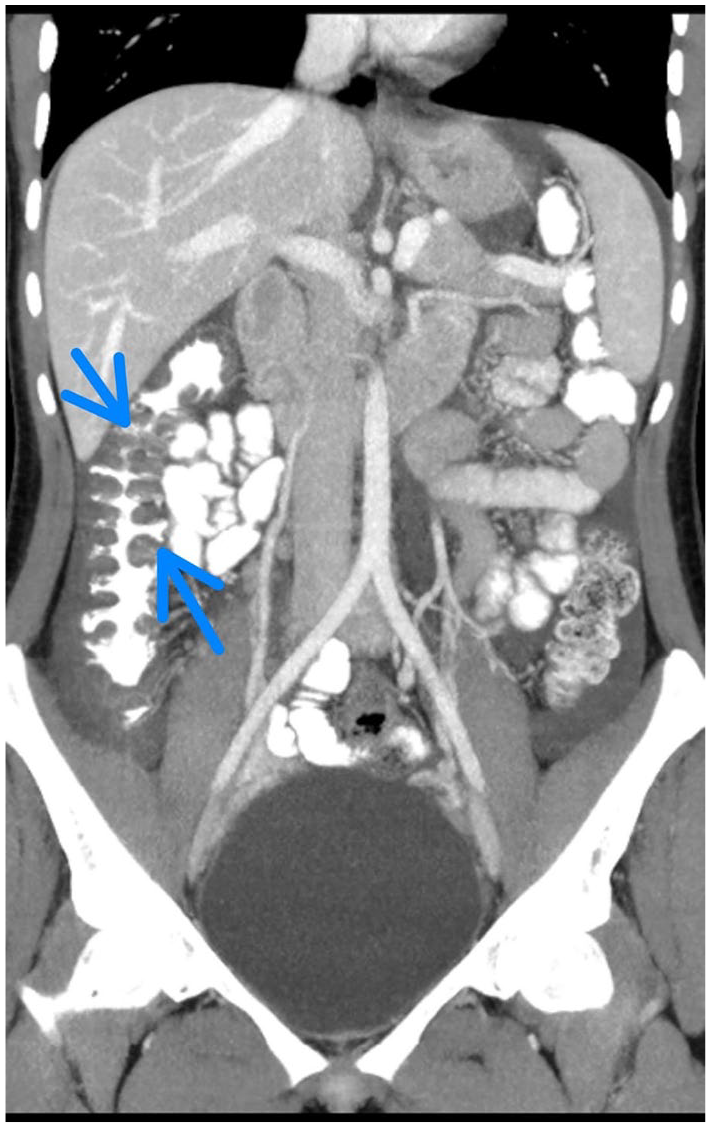
Selected coronal images of oral and intravenous-enhanced abdominal CT show a significant wall thickening with submucosal edema affecting the ascending colon (blue arrows).

Selected axial images of the same study show the affected proximal transverse colon with an abrupt transformation into a normal colon (red arrow).
A gastroenterologist was consulted, who started Cetirizine, an antihistamine, with her as he suspected histamine-mediated angioedema, but no response was noted. The patient was discharged as her symptoms were relieved spontaneously. The family denied doing complement screening before despite the history of danazol taking. So we advise them to do complement screening, as the previous history of angioedema and the long time response to danazol, unresponsiveness to antihistamines, and CT findings, raise the suspicion of HAE being the cause. As these tests do not exist in our institution, they are done in an external laboratory, and we recommend following up until the result comes. After 1 month, results revealed low complement C4 levels of 4 mg/dL (normal range 10–40) an abnormally low function of C1-INH 40% (normal range equals or more than 68%) with normal C1q levels of 187 mg/dL (normal range 50–250). Depending on the result, the patient was diagnosed with HAE. We educated the patient about her condition and advised her to follow up with an allergy and immunology specialist to describe the treatment.
Discussion
HAE, a rare autosomal dominant disorder, stems from C1-INH gene mutations, with 85% of cases showing C1-INH deficiency (Type 1) and 15% having C1-INH dysfunction (Type 2). 2 This causes excessive bradykinin, leading to angioedema flares. 6 HAE, lacking histamine or mast cell pathways, differs from histamine-mediated angioedema, explaining its antihistamine resistance. 7 First attacks often occur in childhood, peak at puberty, and are diagnosed by or after adolescence. 4 Attacks may be triggered by emotional stress, infection, medical or dental operations, etc.; however, some may be idiopathic. 8
Gastrointestinal manifestations of angioedema can include non-specific symptoms, including nausea, vomiting, abdominal pain, and rarely hypovolemic shock. These symptoms mimic an acute abdomen, leading to wrong treatment and unnecessary laparotomies. 5 Abdominal attacks were found to occur in more than 90% of patients. 9 Despite that, it is rarely the main manifestation of HAE. More rarely present is isolated intestinal angioedema, characterized by an edematous swelling of a segment of the bowel with no other manifestation, but it can cause important morbidity. 3 Isolated colon involvement in HAE is quite unusual. 10 which was found in our case. Classically, the symptoms are worse during the first 24 h and then gradually get better throughout the next 48–72 h. 3 This explains the spontaneous relief of our patient’s symptoms.
CT imaging facilitates the diagnosis of the patient suspected of having an abdominal attack due to C1INH-HAE. 9 However, the literature recommends complement screening as an initial laboratory test, with serum C4 being the fastest screening test in emergencies with an accuracy of >95% to exclude C1-INH-HAE. 11 Confirmation of HAE requires a decreased C1-INH function. 4 Although our patient previously responded well to danazol, raising suspicion for HAE, a definite diagnosis was not established due to the patient’s incomplete follow-up with her doctor, and the trial treatment was based on her clinical presentation without genetic or immunological labs. Our patient had low levels of C4, decreased function of C1-INH, and normal levels of C1q, which confirm the diagnosis of HAE. A genetic study is used for sporadic cases and to understand pathophysiology more than diagnosis, which is unnecessary.4,11 Screening of C1-INH serum level is used to distinguish between the types of HAE; Low serum C1-INH levels are diagnostic of type I HAE, and normal levels of C1-INH are diagnostic of type II HAE and both have low C1-INH function. 8
The potential differential diagnoses encompass systemic lupus erythematosus (SLE), inflammatory bowel diseases (IBD), familial Mediterranean fever (FMF), infections, and mesenteric ischemia (Table 1). Lupus enteritis, linked to SLE, shares characteristics like submucosal edema and hypocomplementemia with HAE’s abdominal attacks. 4 The distinction between SLE and HAE lies in their autoantibody and complement profiles. While SLE often involves high antinuclear antibodies (ANA)and low C3 levels, HAE typically exhibits negative ANA and normal C3 levels even during attacks. In IBD or certain infections like Salmonella and Shigella, CT scans often reveal bowel-wall thickening and occasional inflammatory markers, contrasting with HAE’s rare hypocomplementemia and negative inflammatory markers. 4 FMF typically presents with recurrent abdominal pain and fever, while HAE patients are typically without fever. 4 Mesenteric ischemia, caused by various factors, results in sudden abdominal pain and bowel edema with elevated inflammatory markers, unlike HAE. Notably, C1-inhibitor levels are consistently normal in these conditions, highlighting the importance of examining C1-inhibitor levels in cases of abdominal pain with normal C3 levels but low C4 and CH50 levels, as this can help distinguish HAE from other diseases. 4
A comprehensive summary of differential diagnoses for HAE by comparing it with other conditions that exhibit similar symptoms.
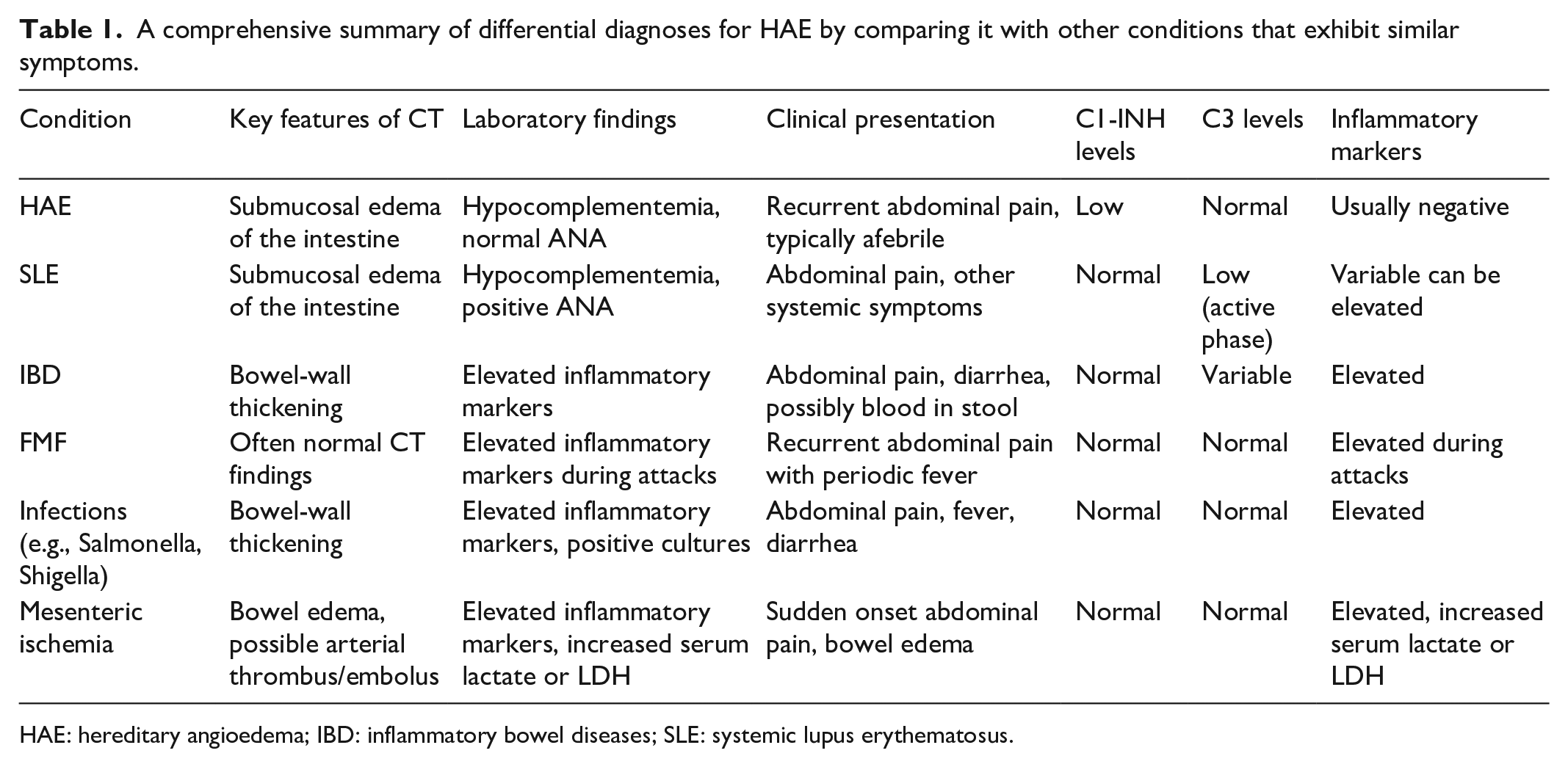
HAE: hereditary angioedema; IBD: inflammatory bowel diseases; SLE: systemic lupus erythematosus.
First-line treatment of bradykinin-mediated HAE attacks includes plasma-derived C1-INH concentrate, recombinant C1-INH, Icatibant, or Ecallantide; however, these drugs are expensive and are not always readily available. Although it is regarded as a second line of therapy, fresh frozen plasma has been utilized to treat acute attacks. Despite all of that the most crucial intervention is patient education, which includes recognizing symptoms and avoiding triggers to prevent acute episodes. Patients should be urged to see the ED immediately if they experience an attack. Use of C1-INH concentrate, lanadelumab (monoclonal antibody to plasma kallikrein), berotralstat (plasma kallikrein inhibitor), danazol, or tranexamic acid is an example of long-term prophylactic therapy.3,12
This case spotlight on another uncommon presentation of HAE emphasizes that a diagnosis of HAE is challenging as HAE has an overlapping presentation and CT findings. 11 Makes it more challenging in our case that it mimics an acute abdomen on presentation without a family history of the same condition and just isolated ascending and transverse colon on CT, resulting in inappropriate treatment and suffering of the patient. Emergency physicians (EP) or gastroenterologists, being unable to recognize this condition results in a delay in the diagnosis of HAE to older age, which increases the risk of mortality not just for the patient but for her blood relevant, as they cannot recognize the attack which may be fatal. 4 To avoid this, the literature recommends screening all family members from all generations for unrecognized angioedema and abdominal pain. 13
Diagnosing and treating hereditary angioedema (HAE) is particularly challenging in underserved and rural areas. A 2015 survey found that about 24% of HAE patients live in rural areas with populations under 20,000, where finding an experienced specialist is difficult. The Centers for Disease Control and Prevention report that rural U.S. counties have fewer specialists, emergency facilities, and transportation options than urban areas. In addition, rural residents are more likely to have government-issued health insurance, which many specialists do not accept.
Riedl et al. identified several challenges in managing HAE in rural areas, including (1) diagnostic barriers misdiagnosis and incorrect interpretation of laboratory results; (2) access to specialist care, travel difficulties, and high costs; (3) local healthcare issues, physician shortages, and delays in therapy initiation; (4) patient education, limited resources for educating patients; (5) medication supply, maintaining adequate on-demand and prophylactic medication; (6) telemedicine barriers, lack of internet access or smartphones; and (7) economic disparities, insufficient insurance, and provider payments. These findings highlight the need for improved strategies to manage HAE effectively in rural settings. 14
We see that by improving healthcare infrastructure and accessibility to essential diagnostics and treatments. In addition, education and awareness among healthcare professionals about HAE are vital to ensure timely and accurate diagnosis, thereby preventing unnecessary treatments and reducing patient morbidity and mortality.
Given that our patient was from a different area, the C1-INH serum level test was difficult to perform in addition to financial issues. So we cannot determine the exact type of HAE, the follow-up process of her treatment and outcomes was restricted.
Conclusion
This case highlights the importance of EP and gastroenterologists’ awareness of such a rare disease with overlapping symptoms and findings on CT, and the importance of evaluating the personal and family history of recurrent severe acute medically unexplained abdominal pain. Educating patients about their condition is more important so they can recognize it and hurry up to the ED. This may help avoid additional misdiagnoses of HAE, delays in receiving appropriate medical attention, unnecessary procedures, and associated complications.
Footnotes
Acknowledgements
The authors express their gratitude to the patient and their family for their great contribution. Also, the authors express profound gratitude to the Polytechnic Medical Students’ Research Association (PMRA) for their invaluable contributions and unwavering support that significantly enriched every stage of the research journey.
Author contributions
M.M. and M.A. contributed to the design of the study and drafting of the manuscript. F.A., O.H.S., and A.M.A. contributed to data collection, data entry, and data interpretation, and contributed to the drafting. M.S. contributed to the design of the study, data interpretation, and supervision of the work. All authors have read and approved the final manuscript. Each author has participated sufficiently in the work to take public responsibility for the content.
Data availability
All data supporting the findings of this study are readily available within the article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.