
Abstract
Multiple intestinal atresia with combined immune deficiency is a severe autosomal recessive disorder caused by the tetratricopeptide repeat domain 7A (TTC7A) gene deficiency, which is characterized by extensive intestinal defects with immune deficiency. This report describes a fetus with TTC7A deficiency who developed meconium peritonitis in utero. Evidence suggests that patients with TTC7A deficiency present with intestinal defects as early as in utero. In this case, intestinal abnormalities were considered during the prenatal examination at week 28, and chromosome and genetic tests were performed. The results indicated that the fetus had a TTC7A complex heterozygous mutation. The male infant underwent surgical treatment after birth and developed severe infection and sepsis, which confirmed the presence of multiple intestinal atresia with combined immune deficiency. Our case suggests an association between meconium peritonitis and the TTC7A gene deficiency, indicating the possibility of severe intestinal defects and immune deficiencies after birth and guiding subsequent fetal treatment choices.
Keywords
Introduction
The tetratricopeptide repeat domain 7A (TTC7A) is a conserved gene that is expressed in a wide range of tissues and organs in the human body, including the brain, bone marrow, testicles, pancreas, ovaries, liver, and blood. 1 In 2013, a TT7CA gene mutation was first detected through whole exome sequencing (WES) in a French-Canadian family with refractory inflammatory bowel disease (IBD)-like diarrhea. 2 TTC7A deficiency is fatal in about two-thirds of patients who show a variety of heterogenous intestinal diseases and immunologic disease manifestations, including IBD, very early onset IBD, multiple intestinal atresia (MIA), intestinal structure loss, and apoptotic enterocolitis, among others. The immune system exhibits severe and varying deficiencies, ranging from mild lymphopenia (ELA) to combined immunodeficiency (CID) and other possible extraintestinal features related to the skin and hair.1,3
Previous studies have reported that TT7CA gene defects are more commonly found after birth due to gastrointestinal and immune deficiency symptoms. The case presented is of a fetus with meconium peritonitis detected by ultrasound during a prenatal examination. Subsequent amniocentesis for genetic testing revealed that the fetus had a TTC7A composite heterozygous mutation, thereby indicating multiple intestinal atresia with combined immunodeficiency (MIA-CID).
Case
The patient was a 29-year-old nulliparous female with no history of adverse pregnancy or childbirth. Both the patient and her husband were in good health and were nonconsanguineous. The pregnancy was natural. The nuchal translucency examination and Down’s syndrome screening in early pregnancy indicated low risk, and there was no difference in TORCH (toxoplasma, rubliiavirus, cytomrgalo virys, herpes simplex virus, others) examination. At 24 weeks of pregnancy, fetal ultrasound indicated the presence of peritoneal fluid. Subsequently, fetal ultrasound at 28 weeks revealed abundant peritoneal fluid accumulation with scattered strong echo, suggesting the possibility of meconium peritonitis in the fetus. The fetal magnetic resonance imaging (MRI) showed that there was no significant defect in the abdominal wall of the fetus and a large amount of irregularly shaped long T1 and long T2 liquid signal lesions were seen in the abdominal cavity, with a local wrapping trend of approximately 3.9 cm × 2.4 cm × 1.5 cm (Figure 1 ). Multiple compartments were seen inside, and the structure of the small and large intestines was unclear, partly disordered, and partly encapsulated. The rectum was shown without dilation, and the remaining colon was unclear. There were no obvious abnormalities in the liver, gastric vesicles, kidneys, gallbladder, and bladder. Diagnosed as “abnormal signal in the abdominal cavity of the fetus, considering fecal peritonitis with the possibility of pseudocyst formation.”

Fetal MRI of the fetus: A large amount of irregular liquid signal foci are seen in the abdominal cavity, with a local trend of encapsulation, ranging from about 3.9 cm × 2.4 cm × 1.5 cm3. Multiple compartments are seen inside.
After the patient and her husband signed the informed consent, the amniotic fluid samples were obtained by amniocentesis, and the peripheral blood samples of the patient and her husband were collected for karyotyping, maternal cell contamination (MCC), WES, and copy number variation analysis.
The American Society of Medical Genetics and Genomics (ACMG) guidelines were referred to for determining the pathogenicity of the variation site. There was a potentially pathogenic variation (NM_020458.4, C.1254C > G, p. Y418, nonsense mutation) inherited maternally and an ambiguous variation (NM_020458.4, C.1577A > C, p. Q526p, missense mutation) inherited paternally, forming a compound heterozygous mutation. Combined with clinical conditions, it was speculated that the compound heterozygous variation of this gene was the genetic cause of the abnormalities observed during the prenatal ultrasound.
A male neonate was born spontaneously at 32 weeks gestation due to premature rupture of membranes. The mother had a low-grade fever (37.7°C) and grade II contamination of the amniotic fluid at delivery, and the neonate developed symptoms of infection after delivery, suggesting that preterm rupture of membranes may have been due to intrauterine infection. The neonate developed an intestinal obstruction and respiratory distress syndrome, had elevated blood inflammatory markers, and was treated with fasting, parenteral nutrition, and antibiotics to fight the infection. On the 10th day of his life, he underwent exploratory laparotomy, intestinal adhesiolysis, abdominal mass resection, small intestinal double tube fistula, appendectomy, abdominal irrigation and drainage, and intestinal atresia of the ileum. Severe abdominal adhesions and pseudocyst formation were observed during surgery. After surgery, he continued to regurgitate after feeding. Two months later, he underwent a repeated exploratory laparotomy, intestinal adhesiolysis, enterostomy retrieval, small bowel T-shaped fistula, and pyloroplasty. Five days after the second operation, the infant gradually started enteral feeding (free amino acid formula powder) intermittently, without regurgitating. However, he continued to present with elevated levels of serum C-reactive protein (CRP), leukopenia, moderate anemia, large stools with mucus and blood, recurrent high fever, and an ultrasound revealed colitis and enteritis. Antibiotics (meropenem and piperacillin) were administered to fight against the infection and supplemented several times with gammaglobulin and other treatments. After 8 months of active treatment without significant improvement, the family chose to stop the treatment, and the infant died at 8 months of age. The results of rectal mucosal biopsy indicated irregular distribution of mucosal glands and atrophy of regional fibers, indicating chronic active inflammation of the rectal mucosa, and a diagnosis of MIA-CID was considered.
Discussion
MIA-CID and meconium peritonitis
MIA-CID (OMIM # 243150) is a rare, severe, autosomal recessive syndrome caused by mutations in the TTC7A gene. 2 Patients with MIA-CID exhibit extensive intestinal atresia from the stomach to the rectum accompanied by CID. Intestinal defects can occur early in fetal development and manifest in utero. 4 Approximately two-thirds of TTC7A gene defects are fatal, with dependence on complete parenteral nutrition, recurrent infections, and intestinal complications being the causes of early death in most patients.1,2,5
The intestinal defect of the fetus, in this case, manifested as meconium peritonitis, which is a sterile chemical peritonitis caused by intrauterine intestinal perforation. 6 The ultrasound manifestations of meconium peritonitis include ascites; scattered calcifications in the abdomen (occasionally extending to the surface of the liver, under the diaphragm, or even to the scrotum); changes in intestinal echo; dilation and thickening of the intestinal wall; and an occasional meconium pseudocyst. There are many causes for perforation, including intestinal atresia, meconium intestinal obstruction, abdominal hernia, intussusception, and volvulus. Susceptibility factors typically include infection or genetic factors. 7 Shiri Shinar et al. 7 evaluated the neonatal outcomes of 244 fetuses with meconium peritonitis. Sixty-seven percent of cases required surgical treatment, the short-term prognosis of newborns was favorable, the survival rate of newborns who required surgery was slightly higher than 90%, and most neonatal deaths were caused by postoperative infections.7,8
Since 2013, more than 50 cases of TTC7A mutations have been reported in the literature and, unfortunately, more than half of these succumbed to the disease, with a median age of survival of less than 12 months. 9
Pathological characteristics of patients with TTC7A defects
The results of the intestinal mucosal biopsy after the birth of the fetus indicated the irregular distribution of mucosal glands and regional fiber atrophy, which was indicative of chronic active inflammation of the intestinal mucosa (Figure 2). Pathological changes in the intestines of patients with TTC7A gene mutations are often characterized by narrowing or complete disappearance of the intestinal lumen, fibrosis and calcification of the intestinal wall, reduction of lymphatic follicles, and thickening of the intestinal wall, indicating chronic enteritis. Histological analysis showed mucosal ulceration, loss of epithelial polarity, mucosal collapse, and loss of apoptotic bodies and villous structures, which are common features in the histology of patients. 10
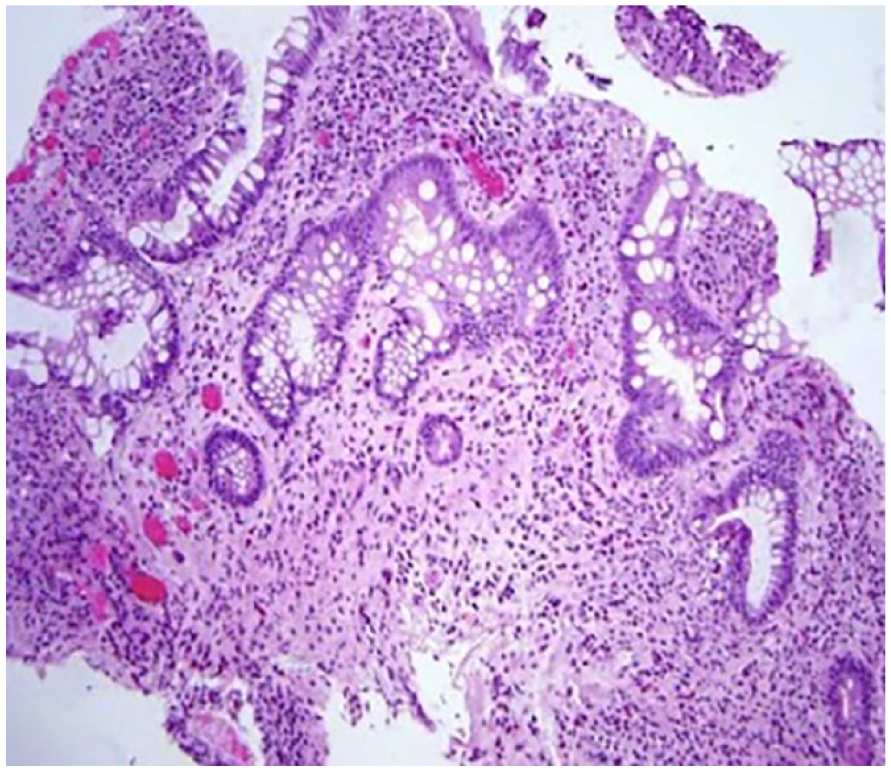
Intestinal biopsy pathology showed irregular distribution of mucosal glands and local fibrous atrophy.
Genotype and phenotype comparison of TTC7A gene mutation
Genotype-phenotype comparison shows that patients with homozygous or compound heterozygous missense mutation of biallelic genes do not affect the tetrapeptide repeat domain of TTCA7 protein and usually have a better prognosis. 11
For example, Woutsas et al. 12 reported two patients, one with a pure missense mutation in the TTC7A gene who showed combined immune deficiency and mild structural intestinal defect, and the other with significant genomic deletion in exons six to eight of the TTC7A gene resulting in a frameshift and premature transcription termination codon. The patient presented with typical MIA-CID.
Despite the previous examples, there are no simplified rules about the relationship between patient genotypes and phenotypes. Mou et al. 13 reported a patient with MIA-CID in which WES of peripheral blood samples showed a pure TTC7A missense mutation; however, the patient exhibited an MIA phenotype. The fetal genotype, in this case, was a compound heterozygous mutation, including a potentially pathogenic nonsense mutation and an unknown missense mutation. Further research is needed to confirm the pathogenicity of this paternal missense mutation.
Conclusion
TTC7A deficiency has been shown to cause intestinal and immune diseases of varying severity. These intestinal lesions can manifest in utero during the fetal stage. In this case, the fetus with the TTC7A gene defect exhibited severe in utero meconium peritonitis as the primary manifestation. This is the first report of a fetus with a TTC7A mutation found during prenatal diagnosis, highlighting the importance of considering genetic problems in fetuses in the context of fetal intestinal abnormalities. Patients with mutations in the TTC7A gene often have comorbid immune problems, which can have a significant adverse effect on the fetus if it undergoes surgical treatment after birth. Therefore, genetic testing is important for determining subsequent treatment options when fetal intestinal abnormalities are detected during prenatal examinations. The TTC7A mutation is autosomal recessive and pathogenic. In this case, the ACMG guidelines classified the pathogenicity of the variant locus as a mixed heterozygous mutation with possible pathogenic variants and variants of uncertain significance without strong pathogenicity. In contrast, the infant presented with severe structural intestinal defects during fetal life and immunodeficiency after birth. Therefore, indicating that further studies are needed to investigate the pathogenicity of these two mutations.
Footnotes
Acknowledgements
We thank the patients and their families family for their role in this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and publication of this article: This work was supported by Hangzhou Science and Technology Bureaugrant number 2021WJCY078.
Ethics approval
Ethical approval to report this case was obtained from * the Clinical Research Ethics Committee of the Hangzhou Women’s Hospital, ((2023)no.058)*.
Informed consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.