
Abstract
Bullous pemphigoid is an acquired autoimmune subepidermal blistering disease that can arise following exposure to systemic medication, referred to as drug-induced bullous pemphigoid. Drug-induced bullous pemphigoid is a rare but potentially serious immune-related adverse event that should be considered in patients with advanced malignancies undergoing immunotherapy, with immune checkpoint inhibitors emerging in particular as a well-documented drug association in drug-induced bullous pemphigoid. We present a 74-year-old female with recurrent metastatic programmed cell death-ligand 1–positive squamous cell carcinoma of the head and neck area who developed drug-induced bullous pemphigoid in the setting of immunotherapy with a novel immunoglobulin-like transcript 4 inhibitor (MK-4830) in combination with pembrolizumab. Treatment with upadacitinib, a Janus-associated kinase-1 inhibitor, was pursued for significantly disabling disease that was recalcitrant to standard therapies and ultimately transition to palliative care. Follow-up at 4 weeks demonstrated good response. This is the first report describing the use of a Janus-associated kinase inhibitor for the treatment of bullous pemphigoid.
Keywords
Introduction
Immune checkpoint inhibitors (ICI) such as those targeted against the programmed cell death protein-1 (PD-1) or its ligand, programmed cell death-ligand 1 (PD-L1), have transformed oncological outcomes and the quality of life of patients with various types of cancer. However, these therapies result in non-specific dysregulation of the immune system and aberrant production of autoantibodies resulting in immune-mediated adverse events (imAR).1,2 Notably, anti-PD-1 and PD-L1 agents have emerged as a significant drug association with bullous pemphigoid (BP).1–3 We report a case of drug-induced BP (DIBP) secondary to immunotherapy with pembrolizumab in combination with immunoglobulin-like transcript 4 (ILT4) MK-4830 in a patient with PD-L1-positive metastatic squamous cell carcinoma of the head and neck (SCCHN).
Case report
A 74-year-old female with a history of recurrent metastatic SCCHN and undergoing active immunotherapy was referred to dermatology for assessment of a new-onset pruritic eruption. She had complex oncological history, including recurrent PD-L1-positive tumour at the base of the tongue and metastatic disease to cervical lymph nodes on imaging. Given pathology finding PD-L1 positivity, she was offered participation in a phase I clinical trial of an ILT4 inhibitor (MK-4830) in combination with pembrolizumab. The patient developed a new-onset pruritic eruption following her fifth cycle of the phase I protocol. She otherwise had a history of malignant melanoma syndrome, including history of two melanomas remotely without recurrence. A complete review of systems was negative. On the upper arms and thighs, there were pink erythematous and oedematous papules and plaques with slight targetoid appearance (Figure 1) despite being previously started on oral prednisone 70 mg/day 10 days prior to her initial assessment. There were no vesicles or bullae, and mucosal surfaces were clear. The constellation of clinical history, drug exposure timeline, and protracted pruritus with urticoid papules and plaques favoured possible DIBP secondary to immunotherapy. A skin biopsy of a representative urticoid papule for histopathology with hematoxylin and eosin staining (H&E) as well as perilesional tissue for direct immunofluorescence (DIF) was obtained and was consistent with BP (Figure 2).

Erythematous and edematous papules and plaques with slight targetoid appearance on the dorsal and lateral upper arms (a and b) and legs (c). There was absence of vesicles or bullae and mucocutaneous involvement.

Histopathology with hematoxylin and eosin staining revealed non-specific but did reveal mixed infiltrate with numerous eosinophils within the superficial dermis and presence of basal vacuolar changes without separation of the epidermis from the dermis at (a) 4× and (b) 20× magnification. Direct immunofluorescence (DIF) was obtained and demonstrated linear staining showing (c) linear IgG and (d) linear C3 at the dermal-epidermal junction.
Immunotherapy was continued for cycle 6 given her overall mild presentation and good response to initial prednisone with tapered to 10 mg per day and topical corticosteroid. Within 24 h of the new cycle of immunotherapy, the patient developed a flare of pruritus and widespread blistering eruption. On re-examination, there were persistent urticoid papules and plaques as well as new tense, uniloculated bullae and scattered erosions with hemorrhagic crust on a background of erythema distributed diffusely to the upper arms and thighs, abdomen, upper chest, and neck (Figure 3(a) and (b)), and there were persistent and development new bullae noted despite prednisone 50 mg per day for 6 weeks (Figure 3(c)). In discussion with her medical oncologist, the patient ultimately elected to transition ongoing efforts to end-of-life care. Given her refractory skin disease despite high-dose systemic and topical corticosteroids and taking into account her transition to palliative care from an oncological perspective, she was initiated on a trial of a Janus-associated kinase inhibitor (JAKi), upadacitinib, at a dose of 15 mg/day following extensive discussion addressing that this would be considered off-label use of this medication and that immunosuppressants are typically considered to be contraindicated in the setting of malignancy. The plan for long-term treatment included eventual transition to dupilumab once approved by insurance, with the goal to optimise her treatment with immunotherapy in the setting of an advanced malignancy. The patient did demonstrate initial response to upadacitinib with resolving urticoid eruption, scattered healing erosions, and no active bullae or mucosal progression on re-evaluation at 4 weeks (Figure 4). Unfortunately, the patient ultimately succumbed to her metastatic cancer 1 month later.
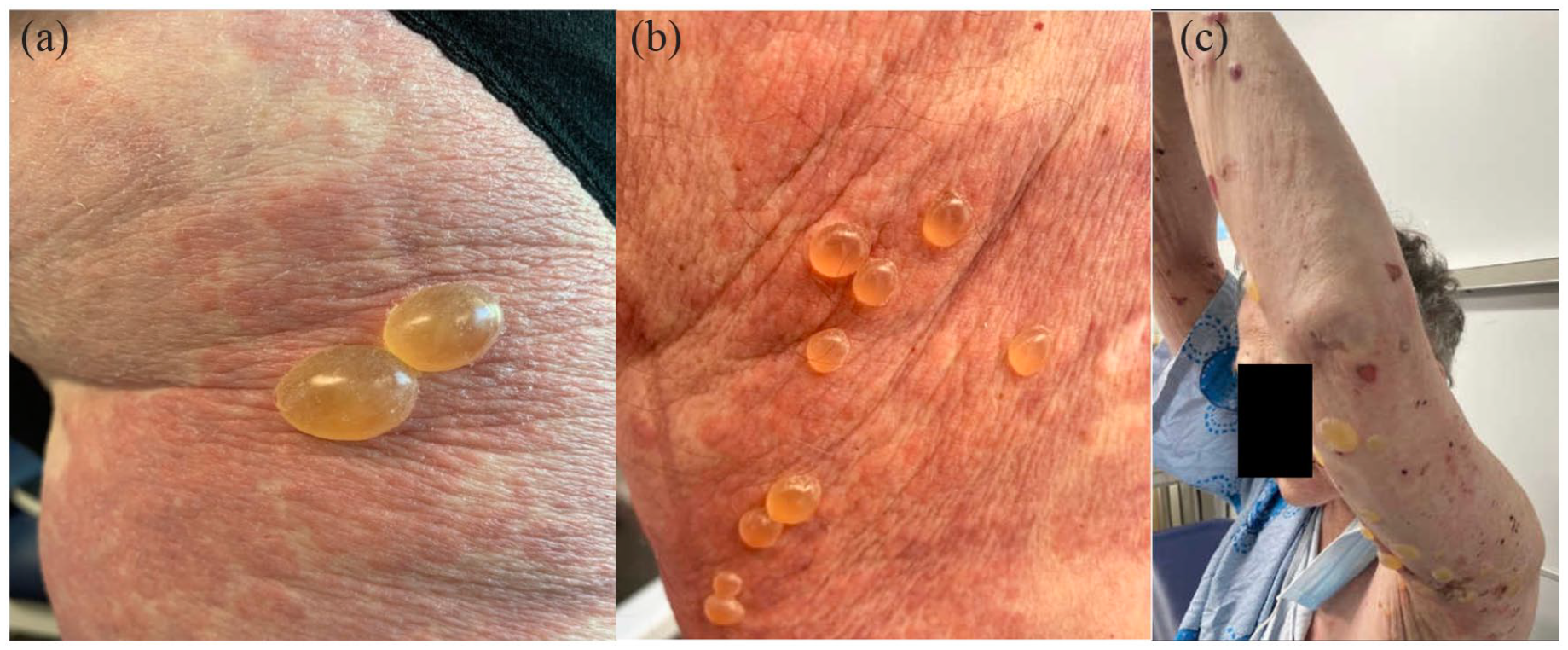
(a) and (b) Numerous tense uniloculated bullae on background of erythema. (c) There were persistent bullae as well as the development of new bullae on follow-up examination, despite prednisone 50 mg per day for over 6 weeks.

Resolving urticoid papules and plaques with few scant bullae and hemorrhagic crusts on the abdomen (a), arms (b and c), and legs (d) following upadacitinib 15 mg per day for 4 weeks.
Discussion
Bullous dermatoses are estimated to constitute 1%–8% of cutaneous immune-related adverse events (irAEs) secondary to immunotherapy with increasing recognition of PD-1/PD-L1 inhibitors as a cause of DIBP. 2 Our patient’s regimen involved a novel ILT4 inhibitor (MK-4830) in combination with pembrolizumab. MK-4830 is a pro-inflammatory agent designed to prevent myeloid cell inhibition of the T-cell response induced by pembrolizumab.
The emergence of DIBP with ICI is well described, with a summary of reported characteristics and perspectives on management reviewed in a recent publication by Muntyanu et al. 1 This case is compatible with other reports of DIBP secondary to immunotherapy in the literature, which have also identified prodromal pruritus or a non-specific erythematous eruption preceding moderate-to-severe skin blistering in up to 87.9% of reported cases.1–3 Similar to our case, the median time between therapy initiation and development of pruritus can be as early as 13–19 weeks, whereas development of classic clinical picture and formation of bullae is often delayed between 28 and 39 weeks, or rarely 78 weeks or later.1–4 DIBP is both debilitating to the patient as well detrimental to the immunotherapy regimen. Early recognition and diagnosis of DIBP in the setting of immunotherapy and advanced malignancies allows for the initiation of appropriate therapy and, as a result, helps to curtail disruption or discontinuation of anti-cancer regimens which are critical in this setting. Similar to our case, treatment discontinuation has been reported in at least 40% of cases of ICI-mediated BP. 3
Several other case studies have reported cancer progression and/or patient death shortly after BP diagnosis and discontinuation of immunotherapy. 4 Our patient had limited response to high-dose systemic corticosteroids but did demonstrate early clinical response to upadacitinib at 15 mg/day. JAKi has a rapid clinical onset between 1 and 4 weeks, which is favourable to achieve early control of disease. There is existing evidence to support therapeutic rationale for the use of JAKi in BP and other immune-mediated bullous diseases, including higher expression of JAK/STAT proteins in skin lesions in BP and other immunobullous diseases compared with healthy subjects, as well as activation of JAK/STAT signalling pathway in the pathogenesis of pemphigoid.5,6 A recent review by Kalantari et al. 7 summarises the use of various JAKi for the treatment of other immunobullous disorders (mucous membrane pemphigoid, dermatitis herpetiformis, and epidermolysis bullosa) which supports the rationale for JAKi as a promising short-term therapeutic option to achieve rapid disease control and help mitigate disturbances in immunotherapy regimens in DIBP. This is the only case to our knowledge documenting use of a JAKi, upadacitinib, for the treatment of BP and offers early support for its therapeutic potential in refractory cases. However, it is important to note that this case describes an exceptional use of upadacitinib in the setting of metastatic cancer in order to provide palliation of the patient’s symptoms in relation to her skin disease with oncological efforts being transitioned to end-of-life care. Immunosuppressant medications and notably JAKi are not recommended in the setting of malignancy, and even certain antibiotics should be used with caution when considering the management of DIBP secondary to ICI.1,8,9 A key limitation includes the inability to observe complete resolution on upadacitinib due to the change of goals of care and ultimately death of this patient as a result of her underlying malignancy. Future studies investigating the relationship between clinical outcomes and the timeline of diagnosis, treatment regimens, and progression of DIBP versus malignancy may provide some guidance in the approach to management in these challenging cases.
Footnotes
Authors’ note
This case has been previously presented at the Canadian Dermatology Association Annual Meeting (June 2022). No material from this conference was published. The manuscript describes original work and is not under consideration for publication by any other journal.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Verbal patient consent from the patient was obtained for publication of the case report content.