
Abstract
Familial hyperchylomicronemia syndrome is a monogenic autosomal recessive disorder that causes severe and refractory hypertriglyceridemia. This uncommon condition is challenging to diagnose and treat and can lead to comorbidities such as acute pancreatitis. Although treatment options are limited in the pediatric population, strict diets and treatments approved for other dyslipidemias may be implemented in familial hyperchylomicronemia syndrome, given the lack of pharmacological interventions available. We report a 14-year-old female presented to the emergency room with abdominal pain suggestive of acute pancreatitis. Biochemical analysis revealed a triglyceride value of 4260 mg/dL. Treatment for triglyceride reduction with a strict CHILD-2 triglyceride-lowering diet, insulin infusion, fibrates, and multiple plasmapheresis were initially insufficient. Primary hypertriglyceridemia was suspected, and genetic testing identified a homozygous pathogenic variant in the lipoprotein lipase gene, diagnosing familial hyperchylomicronemia syndrome. She was discharged with a maximum dose of fibrate, statin, omega-3 fatty acids, and a restrictive diet. At her 1-month and 9-month follow-ups, her triglyceride values were 756 and 495 mg/dL, respectively, without incident complications. Familial hyperchylomicronemia syndrome is an uncommon condition with limited available literature and treatment options, especially in the pediatric population. Acute pancreatitis secondary to severe hypertriglyceridemia is a condition with a high risk of mortality which requires prompt clinical suspicion and treatment.
Keywords
Introduction
Hypertriglyceridemia is defined as triglycerides (TGs) above the 95th percentile for sex and age. 1 Severe hypertriglyceridemia is defined as TGs > 1000 mg/dL, 2 affecting 1:1,000,000 individuals. Severe hyper TG is most often caused by increased presence of TG-rich chylomicrons (CM), 3 which are synthesized after meals by enterocytes for TG transport. 1 The TGs in CM are hydrolyzed by lipoprotein lipase (LPL), a rate-limiting enzyme crucial for TG-rich molecule clearance. LPL hydrolyzes plasma TG into free fatty acids and glycerol and catabolizes CMs into CM remnants.1,4 However, the LPL enzyme is dependent on cofactors and transport proteins, such as the glycosylphosphatidylinositol high-density lipoprotein-binding protein type 1 (GPIHBP1), which is fundamental in keeping LPL anchored to the endothelial cell’s surface, and the lipase maturation factor 1 (LMF1) which encodes a transmembrane chaperone protein critical for LPL maturation.4,5
The etiology of hypertriglyceridemia is classified by primary or secondary causes. 1 The primary causes include genetic conditions such as familial chylomicronaemia syndrome (FCS), which is an autosomal recessive monogenic disorder characterized by a severe elevation in circulating TGs due to mutations involving the LPL gene (95% of cases) or other genes necessary for proper LPL functioning (5%). 2 This syndrome could lead to complications such as acute pancreatitis (AP), associated with high mortality rates. 5 Although timely treatment is necessary to avoid complications, the commonly used TG-reducing medications are not entirely effective in FCS, since they are dependent on optimal LPL function. Emerging treatments such as volanesorsen and lomitapide are limited in low-income countries, 3 especially in the pediatric population, challenging effective treatment and the prevention of complications.
Case presentation
A 14 years and 11-month-old female with an unremarkable personal and family past medical history was admitted to the hospital due to sudden epigastric abdominal pain radiating to the back, with associated vomiting. Upon presentation, the patient was noted to have a normal body mass index (BMI) for her age, no acanthosis nigricans, normal adipose tissue distribution, and adequate puberty onset and progression. Abdominal computed tomography reported necrotizing pancreatitis involving 10% of the pancreatic tissue, and blood work showed TGs at 4260 mg/dL, total cholesterol 518 mg/dL, high-density lipoprotein cholesterol (HDL-C) 5 mg/dL, low-density lipoprotein cholesterol (LDL-C) 108 mg/dL, and amylase 170 U/L (normal range: 30–118 U/L). Upon initial investigation, no secondary causes for hypertriglyceridemia were identified. The patient was not taking any medications and had normal blood glucose, hemoglobin A1c, and thyroid function.
She was admitted to the pediatric intensive care unit, where she received intravenous (IV) hydration, antibiotics, and a crystalline insulin infusion at a rate of 0.08 U/kg/h for 24 h with an inadequate TG-lowering response. Initially, the patient was managed with fasting for 72 h, followed by a restrictive diet of fat after 30% of the total daily caloric intake, 8%–10% total fat per day, <300 mg/day of cholesterol, and limited simple, refined carbohydrates for approximately 15 days. There was insufficient TG response, so the CHILD-2 diet was implemented with a further restriction of saturated fat intake to <7% of calories and a decrease of the daily cholesterol intake to 200 mg, monounsaturated fat <10% of the daily intake, and supplemental water-soluble psyllium fiber. Due to persistently elevated TG levels, medications approved for other conditions were included: gemfibrozil was started and was later replaced by fenofibrate, without an adequate response. Subsequently, eight sessions of plasmaphereses were performed, with an immediate rebound of the TGs to pre-session levels (Figure 1). The patient was stable during her hospitalization, with no additional abdominal pain or AP episodes.
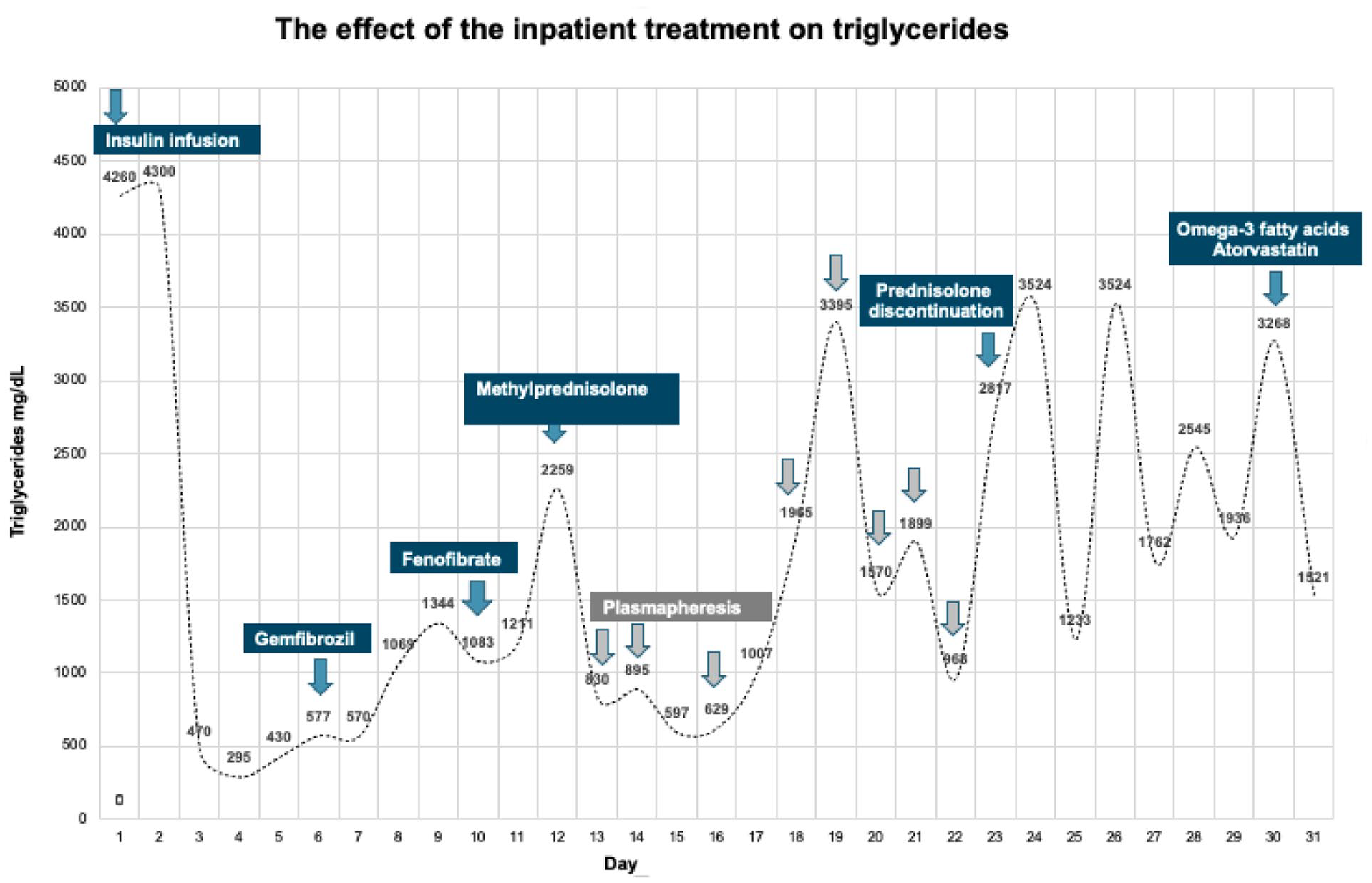
The effect of inpatient treatment on triglycerides.
Further examination revealed hepatosplenomegaly, associated with a speckled pattern antinuclear antibodies (ANA) (1:80) and positive antibodies to extractable nuclear antigens (ENA) (Ro-La-Sm), suggesting a possible autoimmune cause. She empirically received methylprednisolone 1000 mg/day for 5 days, then prednisolone 30 mg/day for 3 days, followed by a taper, with no noticeable reduction in TG levels.
Given the inadequate response to plasmapheresis and empirical glucocorticoid treatment, autoimmune causes were excluded, and primary genetic causes were suspected. A diagnostic scoring system for FCS, FCS SCORE, 1 was applied, and the patient’s score was >10. Exome sequencing identified a previously described pathogenic homozygous mutation in exon 5 of the gene for LPL (c.644G > A).
Treatment with a restrictive CHILD 2 diet along with fenofibrate (200 mg/day), atorvastatin (10 mg/day), and omega-3 fatty acids (4 g/day) BID was prescribed on hospital discharge when the TG level was 1521 mg/dL. At 1-month and 9-month follow-up, the TG values were 756 and 495 mg/dL, respectively (Figure 2). Furthermore, the patient had no recurrent AP episodes and, to date, has not been readmitted. There was no relevant family medical/metabolic history. The patient’s mother and brother underwent a basic lipid profile (total cholesterol, TGs, HDL, LDL) with all values within the normal range; genetic testing could not be performed.

Triglyceride (mg/dL) results after the patient’s discharge: 1-month and 9-month follow-up.
Discussion
In this case, we review the uncommon occurrence of AP secondary to severe hypertriglyceridemia within the context of FCS. This is a rare condition that requires a systematic approach for a timely diagnosis, to provide therapeutic interventions which will reduce morbidity and mortality.
Hypertriglyceridemia in children requires a careful evaluation of secondary causes such as obesity, type 2 diabetes mellitus, nephrotic syndrome, hypothyroidism, pharmacological agents, alcohol use disorder, and autoimmune conditions. Once secondary causes have been effectively ruled out, it is essential to elicit a thorough family history of dyslipidemia, pancreatitis, and premature cardiovascular disease that would raise the suspicion of primary causes with strong genetic components.3,6 In patients with FCS, polygenic and monogenic causes have different presentations. Unlike polygenic causes, such as multifactorial hyperchylomicronemia (MCM), monogenic conditions usually present with fewer metabolic abnormalities but tend to have more pancreatitis. Thus, polygenic causes have a more accentuated and widespread metabolic involvement, and, therefore, the lifestyle interventions required to control the abnormalities are more profound. 7
The pathologic variant discovered in our patient has been previously described in individuals with homozygous LPL deficiency or compound heterozygosity with other pathogenic variants.8,9 The FCS SCORE is a practical diagnostic scoring system with the potential to help to determine which patients require the gold standard genetic testing for pathogenic variants known to cause FCS. 1 This score includes the age of presentation (a younger age confers higher risk), extreme laboratory findings (three consecutive blood tests with fasting TGs > 875 mg/dL or at least one TG value > 1750 mg/dL), a lack of pharmacological response to hypolipidemic treatment, and unexplained recurrent abdominal pain and a history of pancreatitis. 1
Once a diagnosis is made, treatment is focused on lowering the TG levels in order to prevent associated complications. Secondary AP induced by hypertriglyceridemia is associated with TG-rich CMs obstructing the pancreatic blood flow, leading to local ischemia and acidosis. 5 The risk for AP rises for every 100 mg/dL increase in TGs above 885 mg/dL. 2 The development of severe AP with necrosis, like the one presented in this case, may increase mortality by up to 30%. 5
The CHILD-2 diet is recommended for treating hypertriglyceridemia in children.10,11 This diet includes limiting the total daily fat intake to <30%, saturated fat to ⩽7%, cholesterol to ⩽200 mg/day, and monounsaturated fat to ~10% of the daily kcal intake; avoiding trans fats; decreasing simple carbohydrates and increasing complex carbohydrates; and increasing fish in the diet to increase omega-3 fatty acid intake. 10 In addition, dietary fiber like water-soluble psyllium should be encouraged.10,11 Omega-3 supplementation may also be beneficial in those with TGs >200–499 mg/dL. 10 Other available agents include fibrates and statins. However, none of these agents are Food and Drug Administration (FDA)-approved for FCS in the pediatric population. 1 For instance, information regarding the safety and efficacy profile of fibrates such as gemfibrozil and fenofibrate in children is limited.
Novel treatments like the antisense oligonucleotide ApoC3 volanesorsen (which prevents ApoC3 inhibition of TG metabolism and CM hepatic clearance) 7 or the microsomal TG transfer protein inhibitor, lomitapide (which inhibits the intestinal secretion of CMs and liver very-low-density lipoprotein (VLDL)), 8 have recently been used in adult patients with FCS based on non-LPL TG reduction pathways, showing a more than 50% reduction in TGs. 9 However, as with the traditional agents, these novel molecules are not approved in the FCS populations nor in pediatric populations.
In our case, considering the limited approved treatments, we used a regimen based on a restrictive diet along with treatments approved for other dyslipidemias, with an adequate TG-lowering response and no AP recurrence or adverse pharmacological effects during the 9 months of follow-up. The patient received the available TG-reducing interventions indicated for other types of dyslipidemias, such as fasting for 72 h, a strict CHILD-2 low-fat diet beginning on hospitalization day 15, and insulin and heparin infusions, which failed to reduce her TG levels to an adequate range. In addition, she received many plasmapheresis sessions, an available treatment option for rapid TG reduction to reduce the mortality risk associated with necrotizing pancreatitis. However, this option is limited by its high cost, the lack of randomized studies (evidence level C), and the immediate rebound to pre-treatment levels.1,5
The rationale behind this applied regimen was mainly directed at the theoretical reduction of the risk of AP with a multifaceted approach using traditional agents for different types of dyslipidemia accompanied by a low fat diet. Fibrates were specifically chosen, as they enhance LDL-mediated lipolysis by reducing apoC3 levels, activating the peroxisome proliferator-activator receptor (PPAR), and reducing hepatic VLDL production. 9 Furthermore, statins were prescribed as the evidence suggests they can decrease long-term TG levels through VLDL and CM turnover, increase LDL receptor expression to lower cholesterol levels, and delay the onset of atherosclerotic heart disease.12,13 Omega-3 was also indicated as it can decrease CM size and reduce TG by 21%.12,14 It is important to note that despite the theoretical benefit from the previously mentioned medications targeting various lipid metabolism pathways, they have not been proven to work in the FCS population since LPL is non-functional, so any agent that enhances LPL function will probably not entirely work in FCS. Therefore, we cannot conclude that our pharmacological approach by itself caused the appropriate TG control in follow-up. This control was probably mainly due to the restrictive diet.
The treatment goal for patients with severe hypertriglyceridemia is to maintain TG levels under 1000 mg/dL. However, in patients with primary genetic hypertriglyceridemia, it should be lower, at <500 mg/dL, since the postprandial TG concentration can be two to four times greater than the preprandial value. 15 Following an AP event, the TGs should be evaluated (with fasting plasma TG concentration) at least twice a year in well-controlled patients and at least four times a year in patients not meeting the treatment goals. 1
Conclusion
To date, there is no consensus regarding FCS management, and evidence in the pediatric population is insufficient. We describe an adequate response based mainly on a strict diet, accompanied by available medications for different types of hypertriglyceridemia to reduce the AP risk. Further research is necessary to clarify the role, safety, and effectiveness of available drugs that are still not approved in children and to clarify the best interventions to decrease TG levels and prevent AP in FCS.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
Ethical approval for this study was obtained from Fundación cardioinfantil – LA cardio ethics committee and institutional review board (approval No. 33-2021).
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Written informed consent was obtained from the legally authorized representative of the minor subject for the publication of this case report.