
Abstract
Pulmonary nodular lymphoid hyperplasia is a rare, nonneoplastic lymphoproliferative disorder mostly manifesting as one or more nodules or localized lung infiltrates. The lesion comprises reactive germinal centers with well-preserved mantle zones and sheets of interfollicular mature plasma cells, lymphocytes, histiocytes, and neutrophils. The radiological finding is not specific, and the diagnosis of pulmonary nodular lymphoid hyperplasia relies generally on pathohistological and immunohistochemical analyses. The most important differential diagnoses are extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue and immunoglobulin G4–related sclerosing disease. Nonetheless, we present a case of pulmonary nodular lymphoid hyperplasia in a 69-year-old woman with the diagnostic challenge of cytological atypia in alveolar spaces inside the lymphoid tissue, coexisting with the diagnosis of adenocarcinoma of the lepidic pattern. Therefore, this case highlights the importance of identifying these rare benign and reactive lymphoproliferative diseases given the risk of developing not only lymphoma but also carcinoma.
Keywords
Introduction
Primary lymphoid lesions of the lung derive from bronchus-associated lymphoid tissue and pulmonary lymphatic tissue. Lesions involve several nonneoplastic and neoplastic lymphocyte proliferation, and a miscellaneous group of diseases. Neoplastic lymphocyte proliferations include low-grade B-cell lymphoma of mucosa-associated lymphoid tissue (MALT), as well as other non-Hodgkin and Hodgkin lymphomas, while a miscellaneous group of diseases includes posttransplant lymphoproliferative disorders, AIDS-related lymphoma, and intravascular lymphomas. Nonneoplastic lymphocytic proliferation includes reactive lymphomatoid hyperplasia, follicular bronchiolitis, lymphomatoid interstitial pneumonia, and pulmonary nodular lymphomatoid hyperplasia (PNLH). 1
PNLH was initially termed as pseudolymphoma by Saltzstein in 1963, 2 while in 1983 Kradin and Mark 3 described PNLH as one or more nodules or localized lung infiltrates, which consist of a reactive lymphoid proliferation. In the World Health Organization (WHO) histological classification of lung and pleural tumors in 1999, the lesion was recognized and defined as nodular lymphoid hyperplasia of the lung. 4 It was presumed that the lesion might harbor localized clones of lymphoma since no immunohistochemical and molecular studies were performed to exclude a MALT-type lymphoma. Nonetheless, Abbondanzo et al. 5 suggested that PNLH is a rare benign and reactive pulmonary lesion ranging from follicular hyperplasia to diffuse hyperplasia of the bronchus-associated lymphoid tissue. Thus, PNLH was excluded in the latest fourth edition of the WHO classification of tumors of the lung, pleura, thymus, and heart.
We present a case of PNLH in a 69-year-old woman that exhibited clinical and pathohistological diagnostic difficulties. Namely, this unusual and, by definition, benign lymphoproliferative disorder was combined with malignant epithelial lesion, that is, adenocarcinoma of the lepidic pattern, which is very unusual and rare. The case highlights the importance of recognizing the radiological and clinicopathological characteristics of PNLH and considering a surgical treatment, especially when the case is suspicious.
Case report
A 69-year-old woman underwent a computed tomography coronary angiography (CTCA) because of cardiac problems, and by accident, in the upper left lung lobe, a suspicious nodule was found. The patient had no respiratory symptoms and no history of smoking. A chest computed tomography (CT) showed a well-defined, part-solid nodule (PSN), measuring 20 × 19 mm in the superior lingular segment of the left upper lobe, with slightly lobulated contour and interlobar pleural retraction (Figure 1). Thoracic lymph nodes were not enlarged. CT morphology was suspicious for malignant lesions, and bronchoscopy was recommended.

Chest CT shows a well-defined, part-solid nodule in the superior lingular segment of the left upper lobe, with slightly lobulated contour, interlobar pleural retraction (arrow), and air bronchogram (arrowhead).
Bronchoscopy with brushings and transbronchial biopsy were performed twice, 3 weeks apart. Cytological and histopathological analyses of the biopsy samples in both cases did not give positive results. An 18F-fluorodeoxyglucose-positron emission tomography/computed tomography (18F-FDG-PET/CT) revealed a PSN of slightly increased 18F-FDG metabolism. Due to nonspecific results, the multidisciplinary team suggested an open surgical biopsy, which the patient refused. Furthermore, the team recommendation was chest CT follow-up.
After 2 years on the control chest CT, volume progression of 15% was noted. In addition, the 18F-FDG-PET showed volume progression with approximately equal increase in metabolism as before, which is suspicious for slow-growing malignant neoplasm. There were no other new foci of FDG either in the lungs or in the mediastinum that would indicate the progression of malignancy. The patient underwent open lung surgery. A tissue sample was taken from the suspected area for cytological intraoperative analysis. It was a lung fragment of harder consistency of size 23 × 19 mm. The cross section shows a grayish-white area, and a cytological imprint is made.
The intraoperative cytology examination showed clusters of uniformly atypical cells in monolayer sheets and single cells with high nucleus–cytoplasm (N/C) ratio, scant basophilic cytoplasm, anisonucleosis with one or more usually small and occasionally prominent nucleoli, and granular chromatin (Figure 2(a)). In the background, there were lots of little lymphocytes, some plasma cells, histiocytes, and peripheral blood (Figure 2(b)). Occasional bronchial cells with cilia were mixed with the single tumor cells. The cytology picture was not typical for carcinoma, but cells fulfilled the criteria for malignancy. After cytological confirmation of the malignancy, a left lung lobectomy was performed.

(a) Intraoperative cytology of nodular lesion shows clusters of tumor cells of high N/C ratio, granular chromatin, and scant basophilic cytoplasm (May Grunwald Giemsa [MGG] stain, ×400). (b) In the background are seen many little lymphocytes, histiocytes, peripheral blood, and some benign bronchial cells (arrow) (MGG, ×100).
Pathohistological findings in the resected lung showed a well-demarcated mass (Figure 3(a) and (b)) composed of reactive germinal centers, well-preserved mantle zones, and sheets of interfollicular mature plasma cells, lymphocytes, histiocytes, and neutrophils (Figure 3(c) and (d)). Granuloma, necrosis, amyloid depositions, and Dutcher’s bodies were not observed, but interfollicular fibrosis and fibrosis around the alveoli close to respiratory bronchioles could be found (Figure 4(a)). The alveoli were lined by atypical cells (Figure 4(b)). On higher magnification, the pleomorphic cells were focally densely packed showing cell–cell contact (Figure 4(c) and (d)). Immunohistochemical staining with CD20 detected B lymphocytes localized in germinal centers of follicles, and staining with CD3 revealed perifollicular T lymphocytes (Figure 5(a) and (b)). Bcl-2 was absent in the reactive germinal centers, although BCL-2 was noted in the mantle cells and T lymphocytes within and between germinal centers. The heterogeneous reaction was observed in the number of perifollicular plasma cells with CD38 (Figure 5(c)), lambda, and kappa antigens. Staining with IgG4 was seen at low levels in plasma cells (Figure 5(d)). The finding was consistent with nonneoplastic pulmonary lymphoproliferative disorders of PLNH. However, the PLNH contained a focus of parenchymal lesion close to respiratory bronchioles with thickened septa and the alveoli lined with atypical cells, along with the diagnosis of atypical adenomatous hyperplasia bur; according to size, the lesion was diagnosed as adenocarcinoma in situ in association with PNLH.
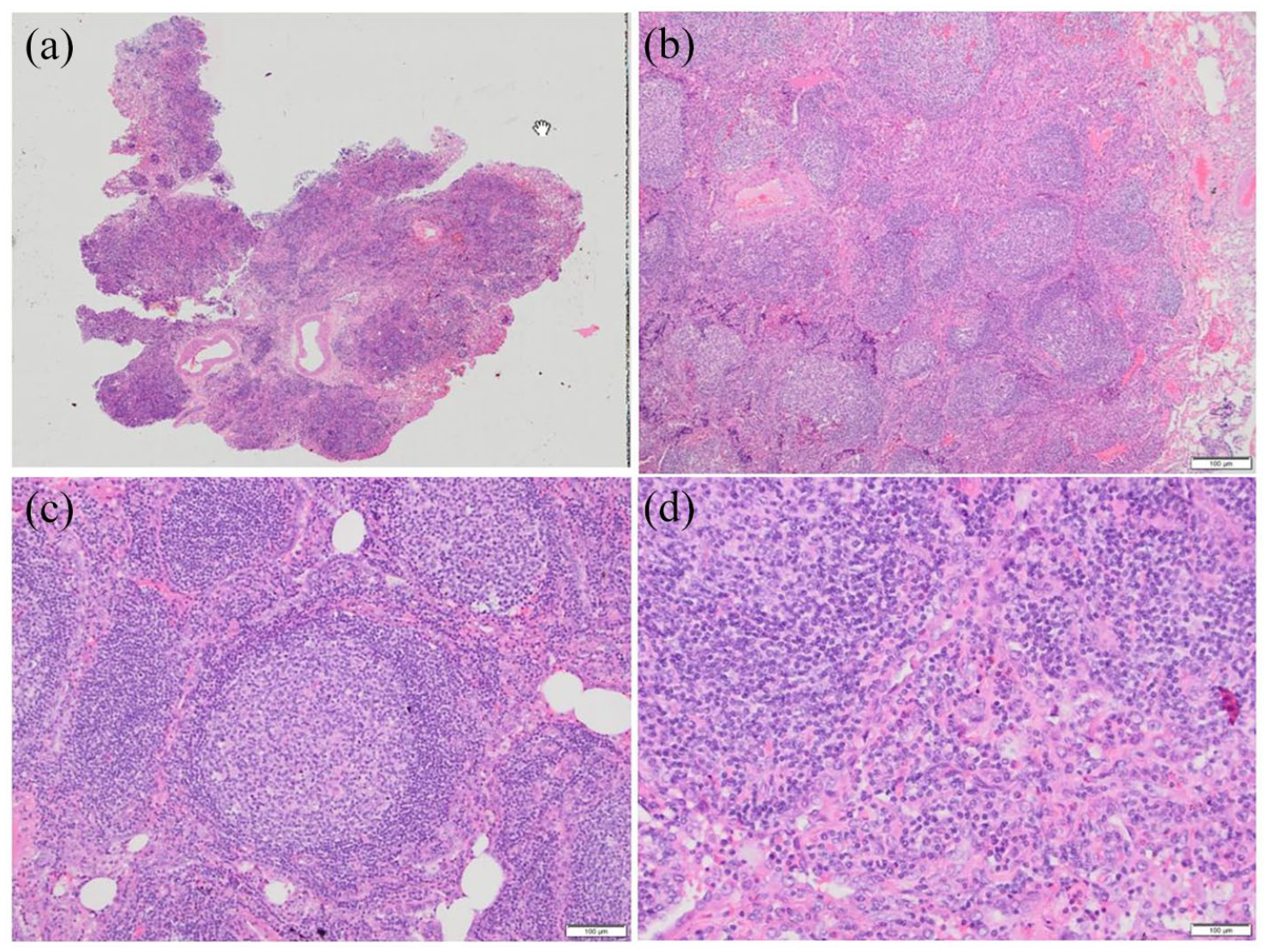
(a, b) Pathohistological staining of the well-circumscribed mass of nodular lymphoid hyperplasia in the lung (c) with reactive well-developed secondary lymphoid follicles. (d) On the higher magnification, germinal center cells, interfollicular lymphocytes, plasma cells, histiocytes, neutrophils, and eosinophils are seen.

(a) Parenchymal pulmonary lesion close to respiratory bronchioles characterized by lymphoid hyperplasia and fibrosis around the alveoli. (b) The alveoli are lined by atypical cells. (c and d) On higher magnification, the pleomorphic cells focally densely packed showing cell–cell contact can be seen.
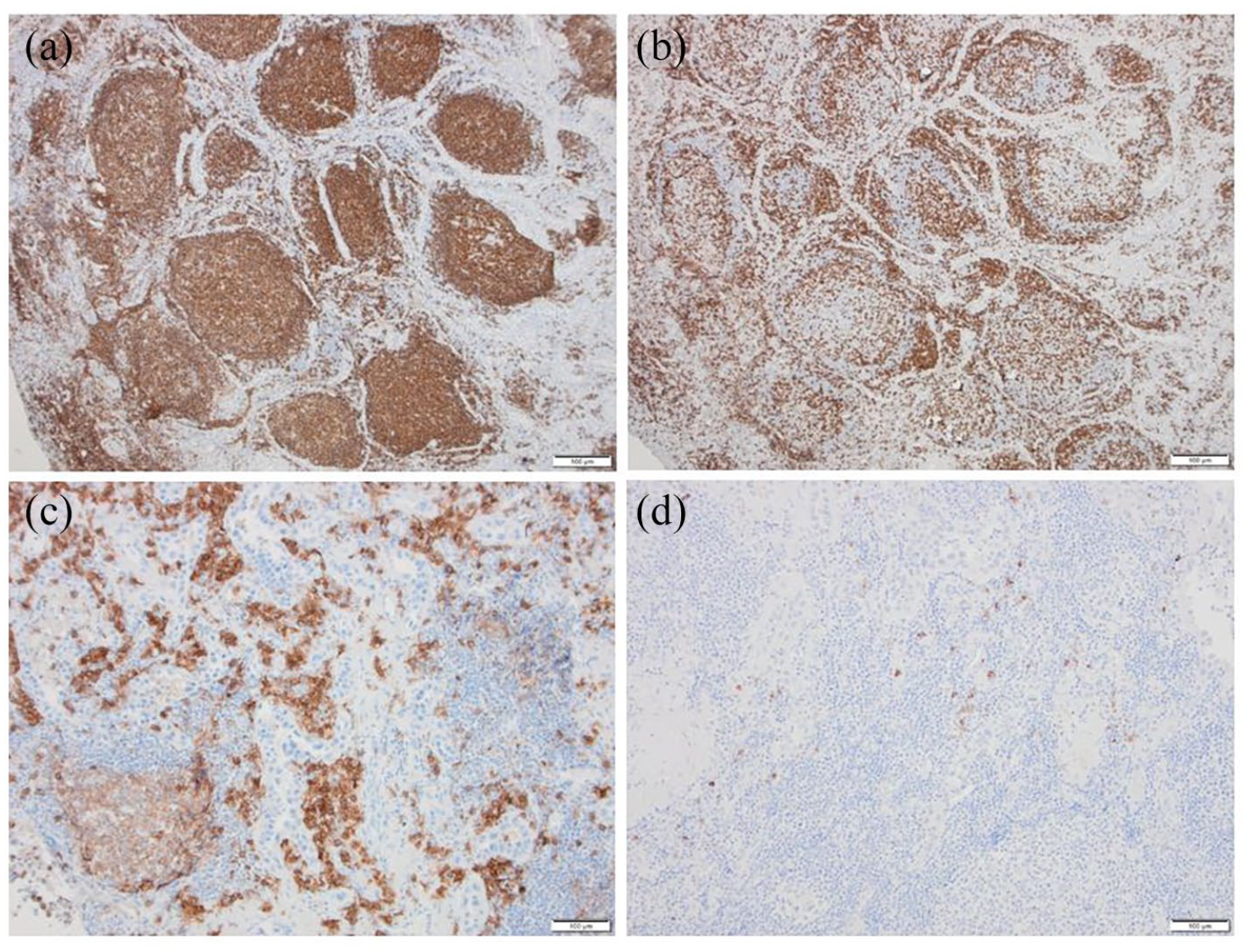
(a) Immunohistochemical staining with CD20 shows tightly packed B cells predominantly within reactive follicles and (b) CD3+ T cells outlining the follicles and throughout the interfollicular space. (c) Plasma cells stained with anti-CD38 are numerous in the interfollicular space, (d) while IgG4+-plasma cells are very rare.
Postoperative recovery proceeds smoothly, and the patient is discharged on the 11th postoperative day. No additional treatment was administered. On control CT scan, after 7 months, a small organized pleural effusion in the left chest is seen, with no signs of spreading malignancy.
Discussion
We present a patient with a single nodular pulmonary lesion that presented preoperative and postoperative diagnostic difficulty. Preoperatively, the differentiation between benign and malignant lesions was challenging. The lesion was followed up for 2 years, and a mild progression was observed, so surgical excision was proposed and performed. Postoperatively, the difficulty to confirm the coexisting epithelial malignancy inside the PNLH, which is considered a rare and benign lymphoproliferative reactive lesion with some risk of malignant transformation to lymphoma within previously polyclonal cell infiltrate, 6 was very challenging.
The lesion in our patient was asymptomatic, detected accidentally during coronagraph analysis, as in the majority of reported cases, although nonspecific respiratory symptoms such as cough, dyspnea, pleuritic chest pain, and hemoptysis have been also described.5,7,8 Most of the earlier reported cases of PNLH have been solitary pulmonary nodules, as in this particular case, but after the first report in 2005 9 multiple nodular lesions were also described.7,10,11
PLNH is usually detected initially through chest radiographs or CT scans. On radiological findings, the lesion appears as a solitary solid or subsolid nodule and occasionally with lobulation, and it cannot be easily distinguished from a malignant lesion, especially when the size increases during follow-up. 8 Particularly the multiple and bilateral ground-glass opacities in the differential diagnosis include malignancy, adenocarcinoma (minimally invasive or invasive), multifocal lymphoma, and multiple focal fibrosis.7,11
On a CT scan, the PNLH can present with multiple ground-glass opacities, indicating the need for differential diagnosis from bronchioloalveolar carcinoma. 7 The PET can also be included preoperatively for the differential diagnosis. 12 In the first reported case, the authors concluded that it may not be useful since the imaging findings of PNLH (including PET-CT) were very similar to those of malignant tumors. 8
In our case, we could not reach a definitive diagnosis with minimally invasive techniques. It was remarked that even an aspiration biopsy might be unhelpful. 8 The diagnosis of PNLH mainly relies on pathological and even immunohistochemical examination postoperatively, as it was in our case. The most important differential diagnosis for PNLH is an extranodal marginal zone lymphoma of MALT and immunoglobulin G4–related sclerosing disease (IgG4-RSD), but it also needs to be differentiated from lymphocytic interstitial pneumonia, lymphomatoid granulomatosis, inflammatory pseudotumor, and inflammatory myofibroblastic tumor.13,14 Despite the similarities, PNLH and MALT lymphomas have different morphological and phenotypic characteristics, so in most cases they can be easily distinguished.14,15 Besides, the radiological findings such as multiple, bilateral nodules, pleural invasion, and prominent lymphangitic extension favor MALT.1,11
Nodular shadow in the lung was also reported to be a major appearance in IgG4-RSD. 16 As a systemic inflammatory disease, it is characterized by the formation of fibrotic lesions containing a lymphoplasmacytic infiltrate abundant with IgG4-positive cells (IgG4+).14,17 Given the presence of heavy lymphoplasmacytic infiltrates in cases with IgG4-related lung disease, previous reports have speculated that at least a subset of PNLH may be one of the various manifestations of IgG-4-related lung disease. 14 IgG4-RSD is characterized by serum IgG4 level higher than 135 mg/dL, numerous IgG4+ plasma cells often greater than 10/high-power field, and the ratio of IgG4+ to IgG+ plasma cells greater than >0.4. 18 On the contrary, most PNLH cases had low IgG4+ cells, 17 as it was in our biopsy.
Differentiation of PNLH from MALT lymphoma or IgG4+-RSD in this case was not a diagnostic problem. However, it was difficult to differentiate reactive pulmonary process from malignant epithelial neoplasm inside the PNLH. It is well known that on cytological specimen the reactive type 2 pneumocyte hyperplasia may be exceedingly difficult to distinguish from atypical adenomatous hyperplasia or carcinoma. 19 It is even described that reactive type 2 pneumocyte hyperplasia may appear more cytologically polymorphic and pleomorphic than bronchioloalveolar carcinoma. 20 In this case, some cytological features such as uniformly atypical cells, nuclei grooves, occasionally prominent nucleoli, and basophilic cytoplasm favor diagnosis of carcinoma. 19 Still, the distinction between bronchioloalveolar carcinoma, atypical adenomatous hyperplasia, and type 2 pneumocyte hyperplasia remains a difficult area in lower respiratory tract cytology specimens. Histological specimen, in this case, allowed the distinction of reactive hyperplasia from atypical adenomatous hyperplasia secondary to alveolar wall thickening and increased the number of alveolar lining cells with atypia. Another differential diagnostic problem was a distinction between atypical adenomatous hyperplasia and carcinoma in situ. Since atypical adenomatous hyperplasia and bronchioloalveolar carcinoma probably represent a continuum of progression of pulmonary intraepithelial neoplasia and the facts that lesion was greater than 5 mm, the diagnosis of carcinoma in situ was done. These cases emphasize the importance of correlating cytological and histologic findings with clinical and radiologic data.
According to the literature available to us, we could not find the case of PNLH associated with small foci of adenocarcinoma in situ except one report of coexistence of lung cancer with IgG4-related disease and obliterative phlebitis in a lung nodule. 16 It was suggested that patients with IgG4-related disease had a higher incidence of malignancies, like lung cancer, pancreatic cancer, colon cancer, and lymphoma.21,22 Tashiro et al. 16 proposed that the production of IgG plasma cells may be a response to unknown antigens.
Little is known about the incidence, etiology, or natural history of PNLH, as it is a relatively rare entity. Our patient did not have a history of other diseases. The lesion has been reported in smokers associated with autoimmune disorders. Lymphoproliferative disorders are assumed to be associated with autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, Sjogren's syndrome, scleroderma, systemic sclerosis, dermatomyositis, polymyositis, Behçet’s disease, and ankylosing spondylitis. The chronic inflammatory process, the history of previous tuberculosis immune dysregulation, and the anti-inflammatory response have been suggested to have a role in the association between autoimmune diseases and lymphoproliferative disorders, also in patients receiving immunosuppressive treatment for autoimmune disease.5,23,24
Most of the previously described cases had single pulmonary nodules, and the patients received complete surgical resection, as in our case, with no recurrence observed. 7 In cases of multiple nodules, only a surgical biopsy was done, and in some cases, the remaining lesions spontaneously regressed during the follow-up period; others remained stable for years or further progressed.7,9,10,25 These cases imply that surgical resection may not be the only treatment for PLNH. Nonetheless, it is very difficult to decide whether or not to remove the nodule(s) because the nature of the disease is poorly understood. More information should be gathered and more cases examined to understand why some patients show progression and others regression.
Conclusion
Our case highlights that although PNLH might be rare, it still exists, and without surgical resection, the diagnosis may be difficult. Besides, postoperatively the differential diagnosis can include MALT-type lymphoma and other benign lymphoproliferative lung diseases, but in our case, the hardest challenge was to confirm the cytological atypia consistent with adenocarcinoma in situ or minimally invasive adenocarcinoma in association with PLNH. Finally, this case highlights that the surgical resection of PNLH was required not only for a definitive diagnosis but also for treatment since observed mild progression was associated with the risk of invasive lung adenocarcinoma.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
Our institution does not require ethical approval for reporting individual cases or case series.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Written informed consent was obtained from the patient for their anonymized information to be published in this article.