
Abstract
Mucopolysaccharidosis is a group of rare metabolic disorders characterized by a deficiency of enzymes in the degradation of glycosaminoglycans. The incomplete degradation process leads to the accumulation of glycosaminoglycans in lysosomes of various tissues, which interferes with cell function. We report three cases that were classified as Hurler—Mucopolysaccharidosis I, Morquio—Mucopolysaccharidosis IV A, and Maroteaux–Lamy—Mucopolysaccharidosis VI. Clinical presentations of these cases vary, depending on each type of enzyme defect. All the patients appeared healthy at birth, and symptoms appear at around 1 or 2 years. Clinical features, radiological findings, and especially enzyme assays have allowed us to establish a definitive diagnosis in these cases. These cases highlight that abnormal clinical symptoms, such as growth failure, coarse facial features, and joint problems, are key points for further investigation relating to mucopolysaccharidosis disease. However, in low- and middle-income countries, it is difficult to have a definitive diagnosis of one of the mucopolysaccharidoses due to lacking enzyme assays.
Introduction
Mucopolysaccharidosis (MPS) is a group of metabolic disorders characterized by a deficiency of enzymes involved in the metabolism of glycosaminoglycans (GAGs) at lysosomal level. The prevalence of all types MPS is one case in 20,000 births.1,2 Different forms of MPS were described separately throughout the 20th century. Clinical presentations of the MPS vary, depending on the type of enzyme defect, the end organ affected, and the glycoprotein accumulated.3–7 The major GAGs are chondroitin-4-sulfate, chondroitin-6-sulfate, heparan sulfate, dermatan sulfate, keratan sulfate, and hyaluronic acid.7–12 MPS can be classified into seven distinct clinical types and numerous subtypes.13–15
The diagnosis could be made through clinical examination and urine, blood, and/or peripheral cell tests.7,16–19 A quick diagnosis is essential because the therapeutics are effective only in patients under 2.5 years of age.3,20–22 Progress in MPS treatment has occurred throughout the world. However, in low- and middle-income countries, it is difficult to have a definitive diagnosis of one of the MPSs due to lacking enzyme assays.
We aim to report three MPS cases in Hue Central Hospital. Clinical, radiological, and biochemical investigations have allowed us to establish a definitive diagnosis in these cases.
Case report
In 2018, our hospital (Hue Central Hospital) received three patients who were clinically suspected of MPS. So, we sent dried blood spots to the Metabolic Laboratory of National Taiwan University Hospital to analyze. With the results of enzyme assays, these cases were classified as Hurler syndrome—MPS I, Morquio syndrome—MPS IV A, and Maroteaux–Lamy syndrome—MPS VI. Here, we report the clinical features, radiology, and MPS-specific laboratory investigations of these patients.
Case 1
A 4-year-old boy had started with the limited movement of his fingers since he was 2 years old. His mother found that he could not lift his arms to his head. He frequently asked for help when he wore clothes. The symptoms worsened, so he was hospitalized. The clinical features included coarse face, joint stiffness, short stature, and mental retardation (Figure 1). He was healthy at birth.

Coarse facial appearance (absence of a fine and sharp appearance of the brows, nose, lips, mouth, and chin).
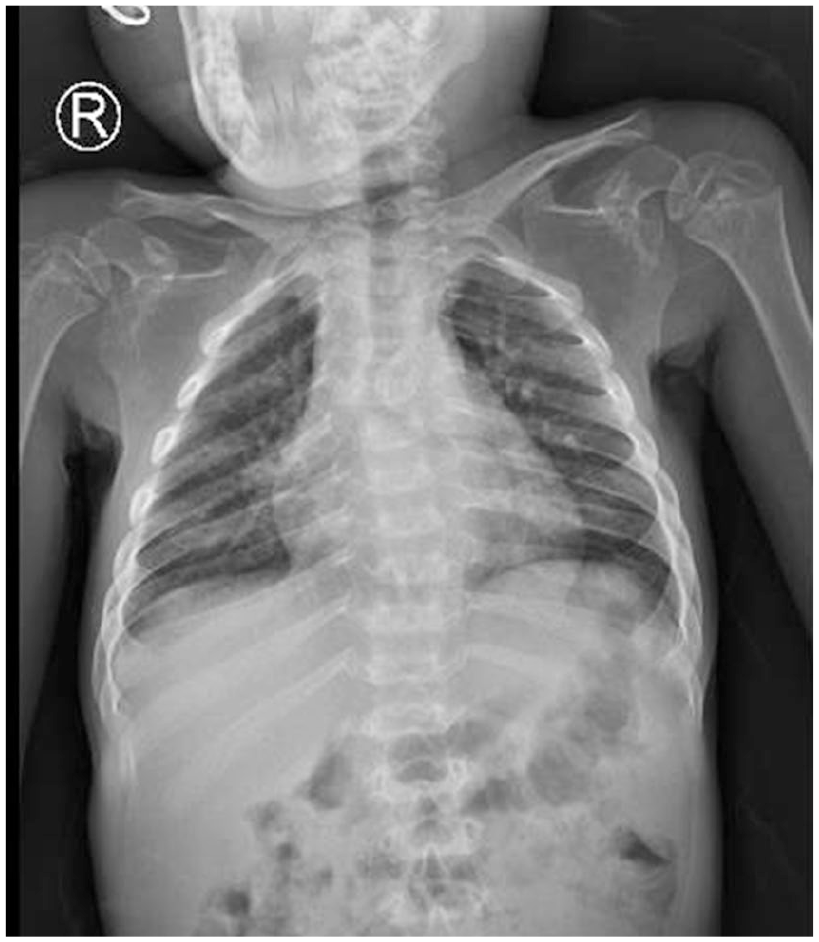
He was 92 cm tall and weighted 13 kg. His hands and feet were large and rather short. Skeletal examination showed short stature with relatively short limbs. The chest X-ray result showed paddle-shaped widened ribs (Figure 2). The respiratory and cardiovascular examination was normal. He had mental retardation. Auditory manifestations and ocular functions were normal.

Paddle-shaped widened ribs on chest X-ray.
A blood test MS/MS assay indicated an α-iduronidase deficiency. This result determined the diagnosis with MPS type I.
Case 2
A 5-year-old girl was admitted to the hospital because of short stature and scoliosis. She was healthy at birth.
Physical examination showed coarse facial features with a wide nose, pectus carinatum, shortened forearm, and genu valgum (Figure 3). The neurological examination verified normal cognitive abilities and behavior. The X-ray image showed the S-shaped scoliosis of the spine (Figure 4).
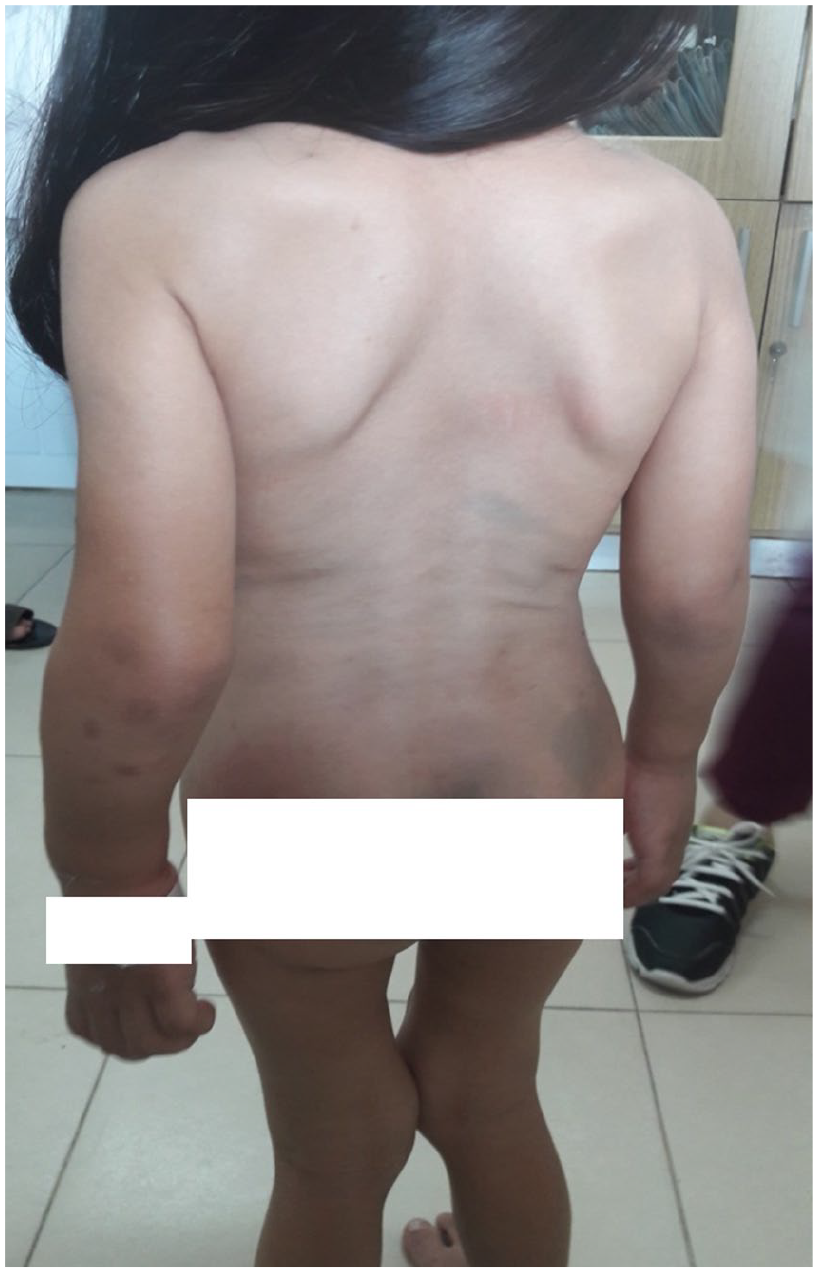
The clinical appearance of the patient with Morquio syndrome (behind view).
S-shaped scoliosis of the spine.
The results of an MPS (MS/MS) assay showed a galactose-6-sulfate sulfatase deficiency. So, this case was classified as MPS type IV A.
Case 3
A 4-year-old girl patient was complaining of finger joint stiffness and had short stature. She was healthy at birth but admitted to the hospital frequently due to respiratory problems.
A physical examination showed the child had a short neck, large skull, rugged appearance, short nasal bridge, wide nose, and swollen eyelids (Figure 5). Her fingers and toes were shorter than normal, and a limited movement in the distal joints due to joint stiffness. Examination of the auditory system showed there was a slight bilateral hearing loss.

The clinical appearance of the patient with Maroteaux–Lamy syndrome.
At first, this patient had normal vision, but after 1 year, she had glaucoma and elevated intraocular pressure (right eye: 30 mm Hg; left eye: 28 mm Hg). She received an operation in 2019.
Echocardiography showed mitral valve prolapse at 1 year after diagnosis with MPS. In hand radiographs, the fingers and metacarpus were short, wide-, and bullet-shaped phalanges (Figure 6). Paddle-shaped widened ribs and thick irregular clavicles were seen in the chest X-ray (Figure 7).
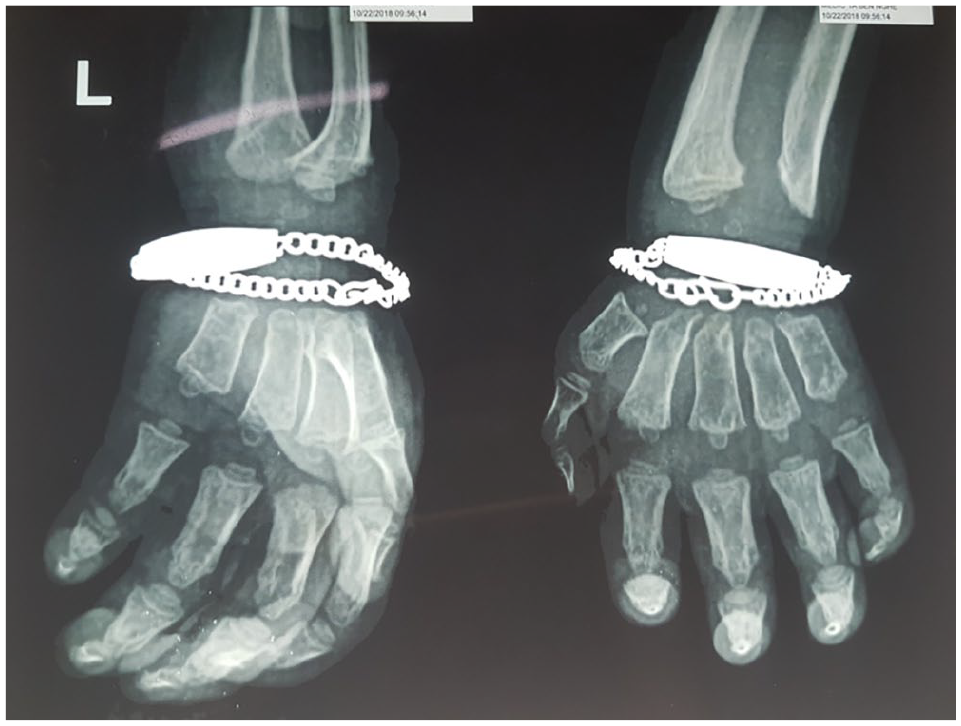
Bullet-shaped phalanges.

Paddle-shaped widened ribs and thick irregular clavicles.
The results of an MS/MS assay showed arylsulfatase B at 0.03 mM/h (normal range: >3.4 assay µM/h). This result suggested an arylsulfatase B deficiency in MPS type VI.
Discussion
Our cases had similar symptoms, such as coarse facial features and abnormalities of the skeleton, especially joint problems. The first and the third patients had joint stiffness, and the second patient had scoliosis. Additional findings included short stature and growth failure. These symptoms made us suspect MPSs. Since the enzymatic assays were not available in our hospital, thanks to a collaboration with the National Taiwan University Hospital, dried blood spots of the patients were analyzed by their Metabolic Laboratory. The results indicated the deficiency of α-iduronidase, galactose-6-sulfate sulfatase, and arylsulfatase B. So, the patients were diagnosed with MPS I (Hurler syndrome), MPS type IV A (Morquio syndrome), and MPS VI (Maroteaux–Lamy syndrome), respectively. Another test to define MPS is the molecular analysis of the related genes, searching for the presence of gene variants, which could be performed in this study.
In our study, all patients were normal at birth, and the symptoms appeared at the age of 1 or 2 years. Most previous reports showed that the onset age usually was around 1 or 2 years, except for MPS VII.14,23,24
Concern to symptoms, in MPS I case, the patient had mental retardation, which was a sign of Hurler syndrome. The mental development begins to regress at about the age of 2.25,26 In contrast to this, in MPS IV A case, the neurological examination verified normal cognitive abilities and behavior. MPS type IV (Morquio syndrome) is also divided into A and B types. In MPS, the central nervous system can be affected.27,28 Most patients with type IV A have normal intelligence. 29 Regarding Maroteaux–Lamy syndrome, it is inherited as an autosomal recessive trait determined by the deficiency of N-acetylgalactosamine-4-sulfatase (arylsulfatase B) activity that leads to incomplete degradation and cellular accumulation of GAG. 30 Our patient was admitted to the hospital frequently due to respiratory problems, which was the key point to make the definitive diagnosis. A recurrent respiratory infection could explain the reason why our patient had slight bilateral hearing loss. Our patient also had mitral valve prolapse and glaucoma, and elevated intraocular pressure. According to Azak and Golda, echocardiography can reveal valvular heart disease with mitral valve 96%, tricuspid valves 71%, and aortic 43%. Therefore, cardiac evaluations are recommended every 1 to 2 years and should include a blood pressure reading, electrocardiography, and echocardiography to assess the cardiac rhythm or conduction abnormality, or changes in the heart structure or function.31,32 Ashworth et al. (2006) showed ophthalmologic features in MPS type VI and noted that they prone to corneal clouding (95%) and glaucoma (50%). 33
The MPS remains an important medical challenge that needs to have an earlier diagnosis as well as prompt intervention. It should be suspected of MPS when patients are presenting with abnormal appearance (short stature, coarse face, short neck, short nasal bridge, wide nose, swollen eyelids, shortened forearm, genu valgum, and coarse face) and with significant clinical symptoms (respiratory symptoms, mental retardation, and eyes and ears problem). These patients need to perform enzyme assays to find a diagnosis.
Conclusion
An MPS diagnosis should be suspected for patients who present with short stature, joint symptoms without signs of inflammation, and other suggestive clinical signs, such as respiratory symptoms, with or without mental retardation. These patients should undergo enzymatic assay to obtain a definitive diagnosis. These were the first three cases we met at Hue Central Hospital. With the collaboration with the oversea hospital that helped us to perform enzyme assays to have the prompt diagnosis
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
Ethical approval to report these cases was obtained from Hue Central Hospital Ethical Committee (approval number: HCH01042018).
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Written informed consents were obtained from the patient’s parents for their anonymized information to be published in this article.