
Abstract
Chronic lymphedema is rarely complicated by an angiosarcoma. Angiosarcoma superimposed on chronic lymphedema (Stewart–Treves syndrome) is usually seen post breast cancer surgery accompanied by lymph node resection of the axilla. This is a case report of a 59-year-old male patient with elephantiasis that developed an angiosarcoma of the lower leg. He died a month after the diagnostic biopsy was obtained. This is a rare multifocal tumor in a male with an unusual lower leg location. We reviewed the literature and the need to differentiate this often deadly lesion from a Kaposi’s sarcoma.
Background/introduction
Angiosarcoma arising in the setting of chronic lymphedema is most often observed in female patients with breast cancer. 1 This presentation is known as Stewart–Treves Syndrome (STS).1–3 It is a rare and almost always lethal entity. The upper extremity is susceptible to lymphedema in females after mastectomy especially with the mastectomy and lymph node dissection. 2 The presence of angiosarcoma is extremely rare in the lower extremities as result of idiopathic chronic lymphedema. 3
Case report
A 59-year-old Caucasian male was referred to our clinic for assessment of his lower leg ulceration and swelling. The patient reported to have been suffering from leg edema for the past 10 years, with apparent deterioration in his condition 3 months prior to his referral. Review of his past medical history revealed chronic heart disease, peripheral vascular disease and ankylosing spondylitis. Physical examination revealed chronic changes of elephantiasis, specifically verrucous skin, with a noticeable serosanguinous foul-smelling discharge, more prominent on the right than the left leg (Figures 1 and 2). Previous computed tomography of his tibia and fibula reported bilateral diffuse skin thickening and abundant subcutaneous fat.

Right leg lymphedema, sero-hemorrhagic drainage, notice extensive fibrinous and eschar tissue (before debridement and antibiotic therapy).

Right leg 1-week after debridement and under broad-spectrum intravenous antibiotic therapy.
Initial treatment included conservative debridement and initiation of intravenous (IV) antibiotics. A 2-week follow-up visit revealed some improvement in the odor and discharge; however, vascular-like lesions were now more obvious on both of his legs (Figure 2), and a punch biopsy was taken. The histopathology report depicted an extensive infiltrate of the dermis with small irregular aggregations of the highly atypical and pleomorphic cells (Figure 3). These atypical cells stained positively for vimentin, CD31 (Figure 4) and, CD34, typical of epithelioid sarcoma (angiosarcoma) but negative for human herpesvirus-8 (HHV-8). Unfortunately, the patient passed away before the histopathological report was available.
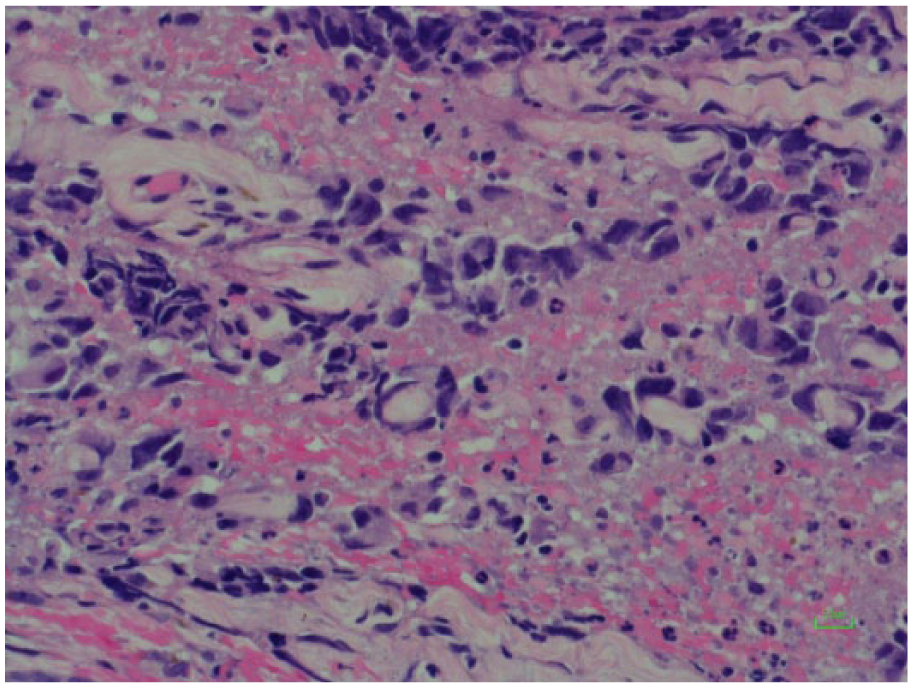
Highly atypical and pleomorphic neoplastic cells form solid aggregations and rudimentary vascular structures (H&E, upper) and strongly express CD31 (immunostaining, lower).

These atypical cells strongly express CD31 immunostaining (lower), and stained positively for vimentin, CD34 typical of epithelioid sarcoma (angiosarcoma) but negative for Kaposi sarcoma–associated human herpesvirus-8 (HHV-8), pankeratin (AE1/AE3), p63, CK7, CK20, S100, SOX10 and CD45.
Discussion
Angiosarcoma arising in the setting of chronic lymphedema is termed STS 1 and accounts approximately 5% of all angiosarcomas. 2 Approximately 400 cases of angiosarcoma related to the STS have been reported in the literature. Most of the reported cases pertain to post-mastectomy patients with concurrent lymph node dissection. However, this procedure, namely radical mastectomy, is rarely performed nowadays; therefore, the incidence of STS has notably declined.2,3 In addition, the axillary lymph nodes are no longer routinely irradiated after lymphadenectomy with the reduction in the associated incidence of chronic lymphedema from 40% to 4%.4,5
The peak age for developing STS is 65–70 years. 6 According to the literature, most of the reported cases involved females7–9 with one report of a male patient with STS after mastectomy. 9 It has also been reported in association with chronically edematous abdominal pannus10–12 and with shunts in renal transplant recipients. 13
STS is a rare clinical entity with incidence that varies between 0.07% and 0.45% of patients treated for breast carcinoma. 14 Lymphedematous changes are required for the development of STS. 15 Sarcomas of the soft tissues account for less than 1% of all the malignant tumors. Angiosarcomas comprise only 2% of all the sarcomas. 16
Massive localized lymphedema (MLL), is a unique presentation of lymphedema, where a large, benign, painless mass, develops in morbidly obese patients. 17 Nine cases of angiosarcoma arising in the setting of MLL have been reported (see Table 1). Interestingly, only four cases (45%) were male patients,18–22 (see Table 1). Not surprisingly, all the MLL patients, as shown in Table 1, are morbidly obese, except our patient. Why chronic lymphedema can lead to angiosarcoma is unclear and controversial. One theory is that chronic lymphedema causes local immunodeficiency, therefore indirectly promoting oncogenesis.23,24 The local immune response within the affected limb is altered by protein-rich interstitial fluid and the lymphatic channels enriched with growth factors, all stimulating lymphangiogenesis and the development of collateral vessels.24–26 According to Ruocco et al., 27 when the local mechanisms of immune surveillance begin to fail, the lymphedematous region becomes an immunologically vulnerable area, predisposed to malignancy including vascular tumors such as STS.
Summary of publications of STS in male patients (including MLL patients).
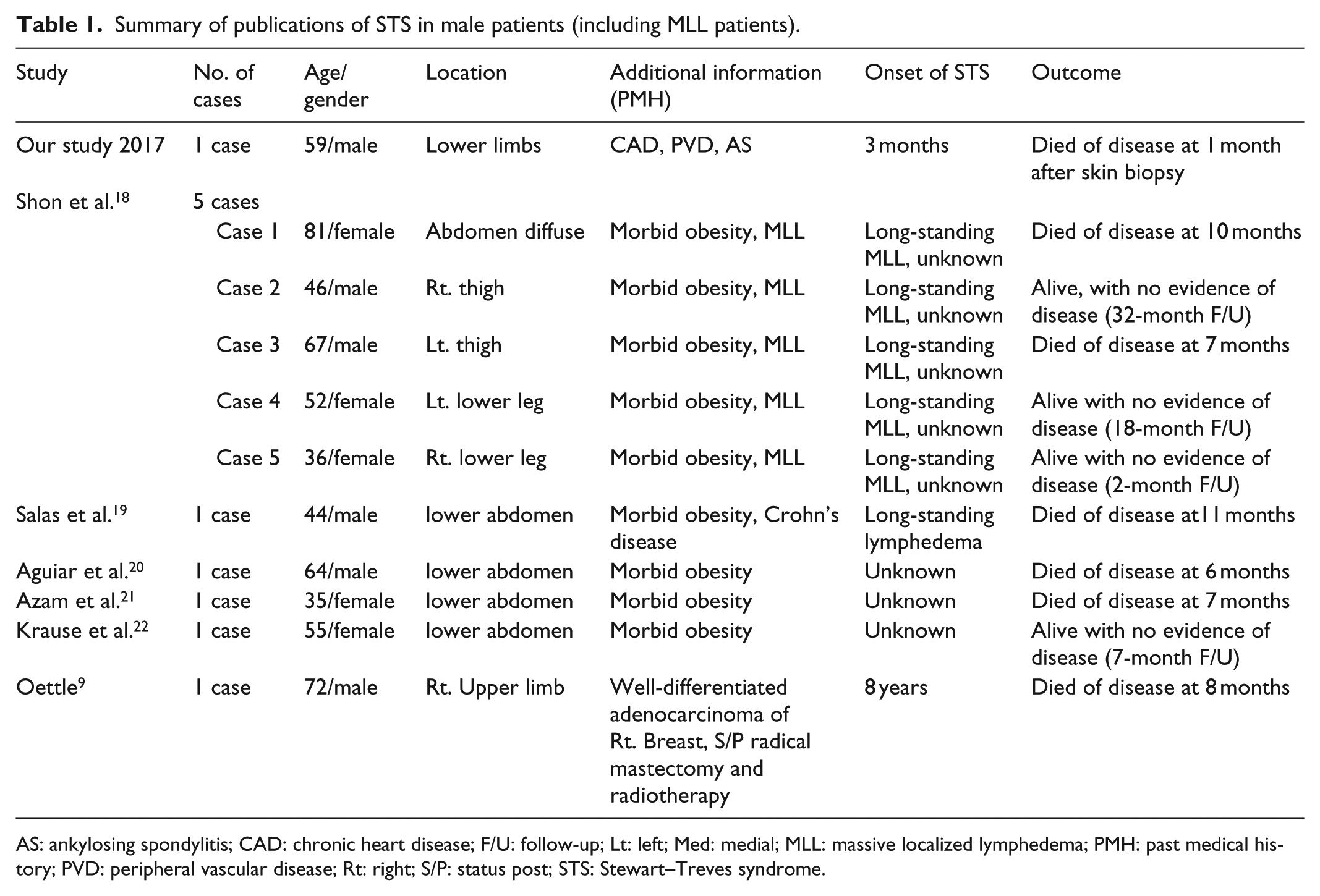
AS: ankylosing spondylitis; CAD: chronic heart disease; F/U: follow-up; Lt: left; Med: medial; MLL: massive localized lymphedema; PMH: past medical history; PVD: peripheral vascular disease; Rt: right; S/P: status post; STS: Stewart–Treves syndrome.
The clinical presentation of angiosarcoma described in the literature usually relates to its common presentation in the upper extremity. This includes initially either a palpable subcutaneous mass or a poorly healing eschar with recurrent bleeding. In later advanced stages, multiple red-blue macules or nodules develop and may become polypoid, with ulceration and necrosis complicating the late stage of some lesions. 28 Lymphangiosarcomas are present in the ipsilateral side in which the radical mastectomy was performed and the lymphedema developed.26,29 Kaposi’s sarcoma (KS) may be somewhat confusing both clinically and histologically, since it can also develop in the presence of lymphedema, although it is not restricted to lymphedematous skin. 30 Immunohistochemical testing for the presence of HHV-8, is the primary way to distinguish KS from angiosarcoma.30,31 In our case, we have also suspected KS; however, immunohistochemical testing was negative for HHV-8.
The management of STS usually combines both surgery and chemotherapy and radiotherapy. 32 However, the role of the latter two regimens is uncertain. Grobmyer et al. reported no significant difference in the survival of patients treated with chemotherapy versus radiation therapy. 33 The best chance of long-term survival is offered with early surgical removal that may include amputation of the affected limb or wide local excision for early disease.32,34 Radical ablative surgery should be performed early in the diagnosis. 34 This is not always feasible, as the tumor is multifocal with an unapparent subclinical spread that makes it difficult to completely excise.32,34–36
However, regardless of the method applied, the overall prognosis of STS is poor, with a high rate of local recurrence and metastatic disease.27,35,36 Angiosarcoma is a high-grade vascular malignant tumor with a mean survival time of 19 to 34 months, despite aggressive combination therapy.36–38 And indeed, most of the patients with STS die from metastatic disease within 2 years.27,28,36–38 Untreated patients usually live 5–8 months after diagnosis.
We have presented a male patient with angiosarcoma of the lower extremity complicating elephantiasis. Our case is unusual not only with respect to the rarity of this syndrome, rather by the unusual lower leg presentation in a male patient. Although a rare complication, it should be considered in patients presenting with chronic lymphedema as early diagnosis may be crucial to improve survival.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Informed consent
Informed consent was obtained to publish the patient information and images.