
Abstract
Advances in sequencing technologies and increased understanding of the contribution of genetics to congenital sensorineural hearing loss have led to vastly improved outcomes for patients and their families. Next-generation sequencing and diagnostic panels have become increasingly reliable and less expensive for clinical use. Despite these developments, the diagnosis of genetic sensorineural hearing loss still presents challenges for healthcare providers. Inherited sensorineural hearing loss has high levels of genetic heterogeneity and variable expressivity. Additionally, syndromic hearing loss (hearing loss and additional clinical abnormalities) should be distinguished from non-syndromic (hearing loss is the only clinical symptom). Although the diagnosis of genetic sensorineural hearing loss can be challenging, the patient’s family history and ethnicity may provide critical information, as certain genetic mutations are more common in specific ethnic populations. The early identification of the cause of deafness can benefit patients and their families by estimating recurrence risks for future family planning and offering the proper interventions to improve their quality of life. Collaboration between pediatricians, audiologists, otolaryngologists, geneticists, and other specialists are essential in the diagnosis and management of patients with hearing disorders. An early diagnosis is vital for proper management and care, as some clinical manifestations of syndromic sensorineural hearing loss are not apparent at birth and have a delayed age of onset. We present a case of Usher syndrome (congenital deafness and childhood-onset blindness) illustrating the challenges encountered in the diagnosis and management of children presenting with congenital genetic sensorineural hearing loss, along with helpful resources for clinicians and families.
Introduction
In the United States, about 3–4/1000 children are born with complete sensorineural hearing loss (SNHL), with 50%–80% of cases due to genetic mutations. 1 Although genetic testing for hearing loss has been available since the 1990s, more recent advancements in DNA sequencing technology (next-generation sequencing) and diagnostic panels have become increasingly reliable and less expensive for clinical use.2,3 Despite these developments, the diagnosis of genetic SNHL still presents challenges for healthcare providers. Inherited SNHL has high levels of genetic heterogeneity, where mutations on different genes or loci can produce the same disease phenotype.4,5 Another complication is that some genetic forms of SNHL demonstrate variable expressivity, in which the same genetic mutation can cause a different phenotype in two different patients, even within the same family. 1
Approximately, 30% of patients with genetic congenital or pre-lingual SNHL have other clinical abnormalities (syndromic hearing loss). In the remaining 70% of these individuals, the SNHL is the only clinical presentation (non-syndromic hearing loss).1,2,6,7 An early diagnosis is vital for proper management and care, as some clinical manifestations of syndromic SNHL are not apparent at birth and have a delayed age of onset. 6 Identifying the cause of deafness can benefit patients and their families by allowing clinicians to offer the proper interventions to improve patient quality of life as well as an estimate of recurrence risks for future family planning.
Although the diagnosis of genetic SNHL can be challenging, the patient’s family history and ethnicity may provide clues, as certain genetic mutations are more common in specific populations. For instance, three types of Usher syndrome exist worldwide (Usher syndrome types 1, 2, and 3) caused by mutations in at least 11 gene loci. 8 Affected individuals typically have a homozygous mutation which is inherited in an autosomal recessive pattern (both copies of the gene have the mutation). The parents of an affected individual usually each carry one copy of the mutated gene, but they do not have any symptoms. Usher syndrome is relatively rare in the general population with an incidence rate of only 4 out of 100,000 births being affected. 8 Communities where consanguinity is more common, may have a higher incidence rate.9–11 A larger proportion of the Louisiana Acadians, for example, have the USH1C c.216G > A which causes the most severe form of the disease, Usher syndrome type 1. This disorder presents with congenital profound bilateral deafness, vestibular areflexia, and retinitis pigmentosa (RP; progressive tunnel vision and blindness) within the first decade of life. 11 Due to vestibular areflexia in Usher type 1 patients, walking is also typically delayed.10,11 Diagnosing Usher syndrome presents its own set of unique challenges in that the blindness due to the RP does not occur during infancy (usually age 10 years for Usher type 1). Early genetic diagnosis of Usher syndrome is necessary to provide the appropriate special educational training programs to manage the loss of hearing and vision. Communication between audiologists, pediatricians, otolaryngologists, and geneticists is essential for the diagnosis and management of these patients. We present a case of Usher syndrome illustrating the challenges encountered in the diagnosis and management of children presenting with congenital genetic SNHL, along with helpful resources for clinicians and families.
Case report
A family with a history of deafness and blindness was referred to the Louisiana State University Health Sciences Center (LSUHSC) Audiology Clinic and the Department of Genetics through the LSU Deaf/Blind Project. The proband (initial patient), 101403, was 17-year-old at the time of examination with profound congenital deafness and recent onset of RP. He was born via vaginal delivery following a normal pregnancy with no clinical history of maternal infections. The newborn hearing screen and subsequent auditory brain stem response (ABR) testing showed profound bilateral hearing loss. At 16 years of age, he developed tunnel vision, followed by rapidly progressive visual impairment and eventual diagnosis of RP. Standard procedure for an infant that has failed the newborn hearing screening is to refer the patient to a pediatric audiologist for a follow-up exam to confirm hearing loss. Families are then informed of the various options to manage the hearing loss, including cochlear implants and training in American Sign Language. 12 In the present case, the patient’s hearing loss was confirmed, and he was trained in American Sign Language. In addition to the current standard of care, we recommend that healthcare providers also refer families with congenital or pre-lingual hearing loss to a medical geneticist to determine whether the cause is genetic. The family had never been referred to a geneticist prior to the visit at LSUHSC and the child had not been diagnosed with Usher syndrome. Both parents have normal hearing. The proband is from a Louisiana Acadian family with six generations of individuals affected with congenital SNHL. A complete pedigree is shown in Figure 1. Twelve family members had deafness only, and 4 also had blindness of a varying age of onset (15–30 years). The family lived in the same geographic area for six generations and consanguinity was reported.

Louisiana Acadian family affected by deafness and Usher syndrome Type 1C.
Methods
Sample collection and DNA isolation
Following our institution’s Institutional Review Board (IRB) protocol, we obtained informed consent from the parents and assent from the minor participant. Peripheral blood was collected from the patient, and saliva was collected from the parents using the ORAGENE saliva sample collection kit (DNA Genotek, Inc., Kanata, ON, Canada). Genomic DNA was extracted using the Qiagen Blood Kit (Qiagen, Hilden, Germany).
Mutation analysis
Since the trio was of Louisiana Acadian ancestry, we tested them for the USH1C c.216G > A at the LSUHSC Genetics research laboratory. Genomic DNA was amplified using polymerase chain reaction (PCR) conditions previously described; PCR products were digested with DraIII (New England Biolabs, Ipswich, Massachusetts, USA) and visualized on a 1% agarose gel. 10 Since the LSUHSC Genetics laboratory is a research facility, results were confirmed using targeted Sanger sequencing at the CLIA-certified Baylor Miraca Genetics Laboratory.
Exome sequencing and bioinformatics
Due to the variable expressivity of the RP in this family, whole exome sequencing (WES) was performed by Personalis Genome Sequencing Center using their ACE Exome™ test followed by bioinformatics analysis. Briefly, whole exome was captured using the SureSelect Human All Exon Kit version 5 (Agilent Technologies, Inc., Santa Clara, CA, USA) on 3 mg of genomic DNA from the mother, father, and proband. Sequencing was carried out with pair-end 100 base reads on the Illumina Hiseq2000 sequencer. This test covered >99.9% of coding bases of these genes at a depth of at least 20×. Data processing and variant calling were performed using the Personalis Pipeline.
Results
Based on the patient’s family history, Louisiana Acadian background, congenital deafness, and progressive retinal degeneration and vision loss, his clinical presentation was suggestive of Usher syndrome type 1. Mutation analysis revealed that the proband is homozygous for the USH1C c.216G > A point mutation that causes Usher syndrome type 1C. The parents were heterozygous carriers of the mutation. Exome sequencing and bioinformatics results were confirmed for the USH1C c.216G > A mutation and identified a variant of unknown significance (VUS) MYO7A c.2798G > A in the proband and the clinically unaffected mother. Additional family members affected with Usher syndrome (who reported not losing their vision until they were 20 or 30 years) and individuals with isolated congenital deafness have also been recruited and are currently being investigated. The pediatric geneticist and genetic counselor provided results and counseling to the family. The patient recently received bilateral cochlear implants to treat his auditory deficits. Aided testing in sound field with bilateral cochlear implant processors indicated hearing sensitivity within normal limits. The patient is receiving professional counseling and learning other communication methods as his vision deteriorates rapidly.
Discussion
The implementation of newborn hearing screening programs and improvements in follow-up services in the United States has dramatically improved the lives of patients with SNHL and allowed for critical interventions at younger ages to preserve speech, social, and emotional development.13,14 Advances in genetic testing in the past 15 years provide clinicians with newer technologies and have reduced the cost of WES and whole genome sequencing, to examine the coding region of the genome and the entire genome, respectively.2,15 The increased understanding of the contribution of genetics toward hearing loss has allowed for the development of clinical diagnostic genetic testing and gene panels for SNHL. Literature evidence shows a shift toward these sequencing technologies to diagnose genetic SNHL.16,17 Despite these advances, the physical evaluation of the patient, obtaining family history, and collaborations/referrals among audiologists, pediatricians, otolaryngologists, geneticists, and other specialists remain vital for diagnosis and management. 18 Upon initial examination of a deaf child, clinical family history including age of onset, severity of the symptoms, affected extended family members, and patient’s ethnicity should be obtained. However, not all genetic cases have a family history of SNHL, since the patient may be the first family member to be affected. Efforts should also be made to distinguish between syndromic (hearing loss with other findings) and non-syndromic (hearing loss only). This difference is not always recognizable, as some features of a syndrome may not manifest until later in life. In total, 50%–70% of genetic SNHL is non-syndromic, with about 40% of these cases caused by homozygous mutations in the gene GJB2, which encodes the protein Connexin 26 and is inherited in an autosomal recessive pattern.19,20 Due to its frequency, the guidelines from the American College of Medical Genetics recommend testing for GJB2 mutations before proceeding to more costly tests such as gene panels or WES/whole genome sequencing. 21 However, mutations in other genes, such as USH1C can cause similar phenotypes. In Usher syndrome, the associated RP-related blindness does not occur during infancy, making it possible to misdiagnose it for non-syndromic deafness. Jervell and Lange–Nielsen syndrome is another syndromic form of hearing loss that can appear non-syndromic in newborns, but is associated with life-threatening cardiac rhythm disturbances (long QT). 22 Furthermore, detailed evaluation by otolaryngology is important for ruling out inner ear bone anomalies as seen in Pendred syndrome. 23 Table 1 provides databases that provide more detailed information about syndromic and non-syndromic SNHL.
Genetic resources for healthcare professionals and patients and families.

A service from the National Institutes of Health.
Despite the advances in next-generation sequencing, genetic SNHL is highly heterogeneous which can complicate diagnosis. There are over 100 genes associated with congenital SNHL. Although the most common mode of inheritance in genetic SNHL is autosomal recessive, autosomal dominant, X-linked, and mitochondrial inheritance have all been documented. Even within the same gene, inheritance patterns may vary, such as in the gene MYO7A in which three separate mutations have been associated with Usher syndrome type 1B, autosomal recessive non-syndromic hearing loss, or autosomal dominant non-syndromic hearing loss.24–26 In the present family, other family members will be screened for the MYO7A mutation to determine their phenotype/genotype correlation. In addition, related individuals with the exact same genetic mutation may express reduced penetrance or variable expressivity. 1
Diagnostic gene panels can uncover pathogenic mutations for autosomal dominant, autosomal recessive, and X-linked inheritance. If mitochondrial inheritance is suspected, a separate test must be ordered. A clinician might suspect mitochondrial inheritance if family history indicates that transmission of the defective allele is through the maternal line. Individuals with mitochondrial mutations may not present with deafness until the administration of aminoglycosides (i.e. gentamycin, tobramycin, amikacin, kanamycin, or streptomycin). 27 The advent of diagnostic panels provides clinicians with a powerful tool to diagnose genetic hearing loss. OtoGenome, for example, tests for at least 86 genes and variants known to cause SNHL of varying severity; however, new genes and mutations are constantly being discovered. A comprehensive, continuously updated database suitable for clinicians and researchers detailing known genes and mutations involved in hearing loss can be found at http://hereditaryhearingloss.org/. 28 Other companies offer their own genetic hearing loss panels in addition to WES and whole genome sequencing services. The cost varies considerably as hospitals have their own pricing agreements with these companies. Genetic tests in addition to a clinical evaluation can help determine whether or not the SNHL is genetic. A genetic counselor will explain the results to the family and provide information regarding recurrence risks and resources for management of the condition. A negative genetic test result, however, does not indicate that there is no genetic etiology responsible for the hearing loss. Diagnostic panels generally cover the coding regions of a gene, as well as splicing junctions, which are where pathogenic mutations are most likely to occur. It is less common, but possible, for mutations to occur in the non-coding regions of the genome which may not be detected by gene panels and exome sequencing. Another challenge is the reporting of variants of uncertain significance (VUS). As more research is carried out, the VUS may be reclassified as benign, likely benign, likely pathogenic, or pathogenic. Table 2 gives a brief overview of the gene panels available for SNHL.
Select diagnostic gene panels for hearing loss.
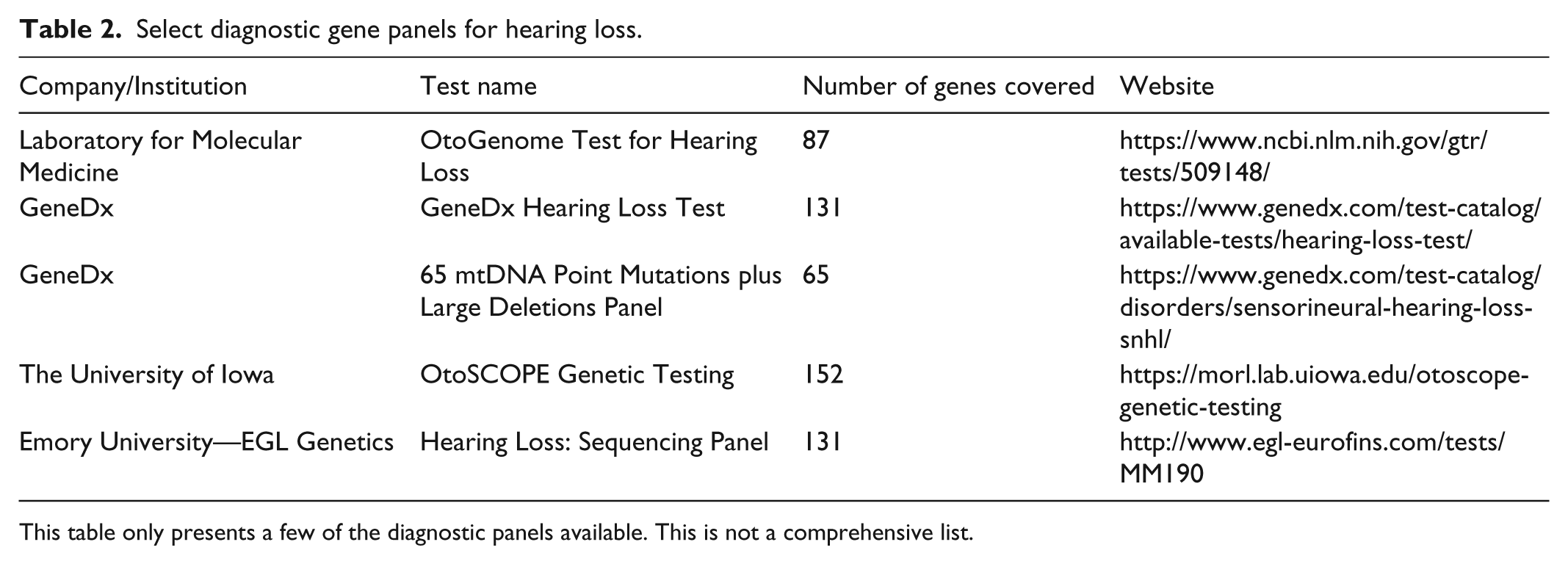
This table only presents a few of the diagnostic panels available. This is not a comprehensive list.
Conclusion
Close-knit communities and founder populations may have higher incidence rates of SNHL.9–11 It is therefore important for clinicians to rule out genetic causes for SNHL, especially if they are serving patients from these particular communities. The presented patient received an initial hearing screen as an infant, but was not followed up by an audiologist, ophthalmologist, or clinical geneticist until the RP had become severe. An early genetic diagnosis would have also benefited this patient in that he would have received interventions tailored to the specific type of hearing loss and impending blindness associated with Usher syndrome. The diagnosis of a genetic cause of deafness can benefit patients and their families, identifying recurrence risks for future offspring and offering the appropriate resources that can improve patients’ communication skills and quality of life.
Footnotes
Acknowledgements
Authors Ayesha Umrigar and Amanda Musso contributed equally toward this publication.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval
Ethical approval to this case report was obtained from Louisiana State University Health Sciences Center New Orleans IRB (IRB# 8434).
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article. Written informed consent was obtained from the minor patient’s legally authorized representative and assent was obtained from the patient.