
Abstract
Background:
The association between smoking dependence and the risk of developing Barrett’s esophagus remains unclear. This study aimed to investigate whether a causal relationship exists between smoking dependence and Barrett’s esophagus, using Mendelian randomization analysis.
Methods:
Two-sample Mendelian randomization analysis was conducted to evaluate the impact of smoking and Barrett’s esophagus. Additionally, we applied summary data-based Mendelian randomization techniques to combine information from genome-wide association studies (GWAS) with expression quantitative trait locus and methylation quantitative trait locus.
Results:
Multivariable Mendelian randomization showed an association between smoking per day (odds ratio = 1.2, 95% confidence interval: 1.038–1.38, p = 0.014) or current smoking (odds ratio = 2.41, 95% confidence interval: 1.06–5.5, p = 0.037) and Barrett’s esophagus. Inverse variance-weighted methods of bidirectional Mendelian randomization also revealed that smoking per day was significantly associated with elevated risks of Barrett’s esophagus (odds ratio = 1.34, 95% confidence interval: 1.092–1.649, p = 0.005), while Barrett’s esophagus was also a susceptibility factor for smoking per day (odds ratio = 1.05, 95% confidence interval: 1.017–1.087, p = 0.003). By incorporating consistent summary data-based Mendelian randomization associations between DNA methylation and smoke/Barrett’s esophagus, gene expression and smoke/Barrett’s esophagus, and DNA methylation and gene expression, we identified that the genetic variant-cg00935895-RBM43 (ENSG00000184898)-smoke/Barrett’s esophagus axis exerted an effect on smoke/Barrett’s esophagus by altering the DNA methylation level, which regulated the expression level of RBM43.
Conclusions:
Our study provides evidence of a bidirectional causal association between smoking and Barrett’s esophagus from a genetic perspective, which sheds new light on the potential role of RBM43 as a mediator in facilitating the impact of smoking and Barrett’s esophagus.
Background
Barrett’s esophagus (BE) is an intestinal metaplasia caused by chronic damage to the esophageal lining and is a known precursor of esophageal adenocarcinoma (EAC).1,2 The current guidelines suggest screening for BE in populations with multiple risk factors. A system is needed to identify individuals within the general population who are at elevated risk of BE, a precancerous condition associated with EAC, to facilitate appropriate endoscopic surveillance. 3
Previous studies have summarized the key risk factors for BE progression and have highlighted promising new risk stratification tools. 4 Studies have also concluded that with the increasing prevalence of cannabis smoking, endoscopic surveillance guidelines may need to be modified for individuals who chronically smoke cannabis. 5 A multicenter study 6 conducted in Europe demonstrated that smoking, even in the absence of gastroesophageal reflux disease (GERD), acts as an independent risk factor for BE. The exact internal mechanisms by which smoking contributes to the development of BE remain unclear, warranting further investigation of the pathophysiological processes involved. Nonetheless, these findings underscore the importance of smoking cessation in mitigating risk, although not entirely in eliminating it.
Building on these findings, our study conducted bidirectional validation using databases from different sources. Furthermore, we aim to provide additional insights into BE by incorporating summary data-based Mendelian Randomization (SMR) analysis. Specifically, we will use whole-blood expression quantitative trait locus (eQTL) and methylation quantitative trait locus (mQTL) data to explore smoking and BE. This approach allowed us to leverage large-scale genomic data to strengthen the evidence base and to address the limitations of previous studies. Recent advances in genetic epidemiology have emphasized the importance of integrating multiomics data, such as eQTL, to better understand the complex disease mechanisms.7,8 By incorporating BE eQTL data, studies have identified candidate genes whose expression is regulated by risk variants in BE-relevant tissues and have facilitated the elucidation of BE pathophysiology. 9
BE is associated with chronic GERD 10 and socioeconomic status, such as smoking, 11 alcohol consumption, and BMI. 12 However, smoking habits that increase the risk of BE warrant further investigation.
Previous findings hold profound significance for screening patients with BE who may be at risk of deterioration, as they can provide timely medical interventions to reverse the condition. 13 Physicians may also benefit from additional BE education and better methods of risk stratification. 14 Thus, regarding smoking behavior, it is feasible to identify individuals who are most at risk of developing BE. Moreover, acknowledging the risks and implementing suitable measures can contribute to the prevention of BE, thus mitigating the associated health burden.
Methods
Study design
Multivariable Mendelian randomization (MR) analysis was used to investigate the causal relationship between five smoking habits and BE, and univariable MR analysis was performed to investigate the results of multivariable MR. Representative phenotypic single-nucleotide polymorphisms (SNPs) were selected as instrumental variables 15 (Figure 1(a)). To serve as effective instrumental variables in phenotype studies, SNPs must satisfy three key criteria: first, they must exhibit a robust association with the phenotype in question; second, they must remain independent of any confounders that could influence the relationship between exposure and outcome; and third, there should be no direct causal pathways between the SNPs and the outcome. 16

Flowchart of the study design. (a) MR analysis; (b) data sources utilized and the conduct of the MR analysis; and (c) summarizes the three-step SMR analysis.
The information used in this study was sourced from publicly accessible databases that had previously received ethical clearance from their original studies. Consequently, no further ethical approval was required for this analysis. However, to ensure the validity of the MR approach, it is critical that the study population be accurately phenotyped. Therefore, we reviewed the diagnostic procedures and ensured that the diagnostic tests for BE were uniformly applied across both BE patients and non-BE controls to minimize the risk of false negatives and other bias. The details of the study design are given in Figure 1. The inclusion criteria for participants were strictly defined to ensure comparability between the groups. Additional sensitivity analyses were conducted to account for the potential bias caused by incomplete phenotyping. Our analysis was conducted in January 2024, encompassing data processing, statistical analysis, and interpretation.
Data sources
Genetic instruments for smoking and BE were selected from summary statistics of genome-wide association studies (GWAS) meta-analysis performed on data from the UK Biobank (https://www.ukbiobank.ac.uk/) 17 and the OpenGWAS database (https://gwas.mrcieu.ac.uk) in European samples, which is an open source, open database, through strict quality control. 18 Details of the data sources are given in Figure 1(b). Additionally, only individuals of European ancestry were included to minimize population stratification effects.
Genetic instrumental variables selection
A threshold of p < 5 × 10 5 was used for genomewide significance in this study. Accordingly, we identified SNPs that showed a strong association with each dietary pattern using this criterion. To ensure the independence of these SNPs, linkage disequilibrium (LD) clumping was conducted using specific parameters (r2 < 0.001 and a window size of 10,000 kb). Additionally, the F statistic was computed to evaluate the potential bias introduced by weak instrumental variables. 19 An F-statistic greater than 10 signifies that the influence of such bias can be considered negligible. 19 The exposure and outcome data were then integrated and standardized according to the effect alleles. SNPs linked to outcomes were excluded from the combined dataset.
Heterogeneity and pleiotropy analysis
Cochran’s Q test was used to detect heterogeneity, with a significant p-value indicating the presence of variation among studies. To evaluate horizontal pleiotropy, MR-Egger intercept analysis was conducted. Additional sensitivity analyses were performed to confirm the robustness of the findings. A p-value for Cochran’s Q statistic less than 0.05, it suggested the presence of heterogeneity. Forest plots and leave-one-out analyses were used to identify any single SNP that may have introduced bias into the results. After eliminating the identified outliers, the MR analysis was repeated to ensure accuracy.
MR analysis
The inverse variance-weighted (IVW) method served as the principal approach for evaluating the causal relationship between dietary habits and the risk of esophageal diseases. Complementary methods, including the MR-Egger and weighted median analyses, were also used. Multivariate MR analysis was conducted to control for potential confounders among various smoking patterns. MR findings were reported as odds ratios (ORs) and 95% confidence intervals (CIs). A two-sided p-value less than 0.05 was deemed indicative of statistical significance. All analyses in this study were performed using the “TwoSampleMR” package (version 0.6.7) in the R software (version 4.4.1, it is supported by the R Foundation for Statistical Computing, which is a non-profit organization based in Vienna, Austria. For more detailed information, please visit the official R website at https://www.r-project.org/).
Gene expression and eQTL, and mQTL dataset
eQTL analysis is the most common approach to evaluate the effects of SNPs on gene expression. 20 The latest version of Genotype-Tissue Expression (GTEx) hosts data for 54 tissues obtained from 948 donors, totalling 17,382 samples. 21 For this project, we obtained fully processed, filtered, and normalized gene expression matrices (in BED format) for each tissue using GTEx version 8 (v8). The gene expression and eQTL data used in this study were retrieved from the GTEx v8 database (https://gtexportal.org/home/). The mQTL data were a meta-analysis 22 of blood mQTL from the Brisbane Systems Genetics Study (n = 614) and the Lothian Birth Cohorts of 1921 and 1936 (n = 1366).
In summary, the SMR methodology leveraged the concept of MR by integrating GWAS, eQTL, and mQTL summary statistics. This approach aimed to assess the pleiotropic associations between gene expression, DNA methylation, and smoke/BE, stemming from a potentially causal variant shared within a locus. The Heterogeneity in Dependent Instruments (HEIDI) test was used to evaluate the homogeneity of these associations. Rejection of the null hypothesis (HEIDI p < 0.05) suggests that the observed association could be attributed to two genetically distinct variants with significant LD.
We employed standard configurations within the SMP framework, such as a minor allele frequency exceeding 0.01, while omitting SNPs in extremely strong LD (r2 > 0.9) with the primary QTL. Additionally, SNPs with weak or negligible LD (r2 < 0.05) with the leading QTL were excluded. Multiple comparisons were adjusted using the False Discovery Rate (FDR) method. The study design and data sources for the three-step SMR are given in Figure 1(c). Data cleaning and statistical analysis were performed using R version 4.4.1 (https://www.rproject.org/).
Investigating overlapping genetic variations potentially linked to multiple traits is the focus of colocalization analysis. By combining GWAS results with molecular QTL datasets using techniques such as SMR or co-localization, researchers can enhance the identification of possible causal SNPs that operate through specific biological pathways. 23 This approach evaluates whether a common causal variant influences the two traits within the same genomic region. To assess shared genetic effects between traits, the “coloc” R package was employed, setting a threshold of PPH4 greater than 0.5 to determine significant associations. 24
Results
Instrumental variables for smoking factors and BE outcomes
Overall, six smoking-related GWAS datasets and three BE-related GWAS datasets were included in our analysis (Figure 1). Detailed information regarding these data is shown in Figure 1.
Multivariable MR analysis
We lack information on which smoking behaviors, including exposure to passive smoking environments, are linked to the outcome of BE. Therefore, we performed a multivariable MR analysis based on five causal associations using the IVW method. The association between cigarette smoking per day (OR = 1.2, 95% CI: 1.038–1.38, p = 0.014) or current tobacco smoking (OR = 2.410, 95% CI: 1.06–5.5, p = 0.037) and BE remained significant in the multivariate MR analysis. However, the remaining three smoking statuses were no longer significant (p > 0.05; Figure 2).

IVW-methods of multivariable MR analysis of smoking status on BE.
Bidirectional MR analysis
In the forward MR analysis, 87 SNPs were extracted, with cigarettes smoked per day as exposure and BE as outcome (Table S1). The results showed IVW (OR = 1.15, 95% CI: 1.013–1.303, p = 0.031), and weighted median (OR = 1.233, 95% CI: 1.013–1.501, p = 0.033; Figure 3 and Table 1). MR results using other validated BE GWAS datasets (ebi-a-GCST90000515) showed IVW (OR = 1.342, 95% CI: 1.092–1.649, p = 0.005; Figure 3 and Table S2). Moreover, reverse MR analysis also showed the BE effect on cigarettes smoked per day using the IVW (OR = 1.051, 95% CI: 1.017–1.087, p = 0.003) and weighted median (OR = 1.08, 95% CI: 1.032–1.12, p = 0.001) methods (Figure 4, Table 1 and Table S3).

Forward MR results between cigarettes smoked per day and BE. (a) Scatter plot depicting the effects of SNPs on BE (ebi-a-GCST003740); (b) funnel plots comparing IVW and MR-Egger methods for assessing bias; (c) leave-one-out sensitivity analysis; (d) scatter plot depicting the effects of SNPs on BE (ebi-a-GCST90000515); (e) funnel plots comparing IVW and MR-Egger methods for assessing bias; (f) leave-one-out sensitivity analysis.
The bidirectional MR estimates for the relationship between genetically instrumented smoking dependence and BE.

BE, Barrett’s esophagus; CI, confidence interval; IVW, inverse variance-weighted; MR, Mendelian randomization; OR, odds ratio; SNP, single-nucleotide polymorphism.
Bold fonts indicate statistically significant differences.
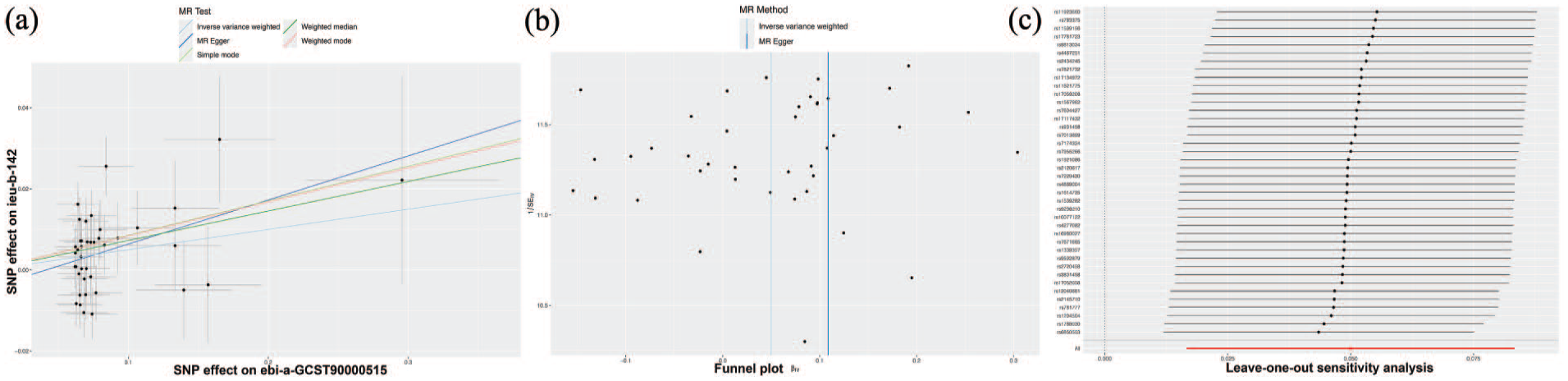
Reverse MR analysis of BE (ebi-a-GCST90000515) on cigarettes smoked per day (ieu-b-142). (a) Scatter plot displaying the effects of SNPs associated with BE; (b) funnel plots comparing results from the IVW and MR-Egger methods; (c) leave-one-out sensitivity analysis summarizing the causal effects of BE-associated SNPs on smoking.
Integration of GWAS and smoking‑related cis-QTL data from whole-blood
We identified 81 genes showing smoking associations from the GWAS datasets (dataset: UK Biobank_22506_111; SMR FDR < 0.05 and HEIDI p > 0.05) (Tables S4 and S5). Figure 5(a) shows the eQTL results for the probe ENSG00000184898 tagged with RBM43 on chromosome 2 (Figure 5(a)). Figure 5(b)–(d) shows that the SNP signals associated with RBM43 were significant across the data from smoking GWAS, eQTL, and mQTL studies. The RBM43 expression level was negatively associated with smoking (beta SMR = −0.034). DNA methylation levels by probe cg00935895 showed a positive effect on smoking (beta SMR = 0.464) and a negative effect on RBM43 expression (beta SMR = −0.303). Taken together, our results suggest a putative mechanism wherein a lower DNA methylation level at the enhancer region of RBM43 upregulates the expression of RBM43 and subsequently decreases smoking risk (Figure 5).

Locus plots showing the significant gene RBM43 and the SMR effect plots. (a) Locus plots display eQTL results for the probe ENSG00000184898 tagging RBM43 within chromosome 2; (b) SMR effect on gene expression and smoking; (c) SMR effect on methylation and smoking; and (d) SMR effect between methylation and gene expression. The x-axis represents cis-QTL effect sizes, while the y-axis represents GWAS/eQTL effect sizes.
Integration of GWAS and BE‑related cis-QTL data from whole-blood
After SMR analysis of BE associations from the UK Biobank_K11_BARRET dataset (Table S6), we obtained six intersection genes with the UK Biobank_22506_111: ITIH4, MUSTN1, TMEM110, HEMK1, RBM43, and RP11-263K19.4 (Figure 6(a)). As a result, only RBM43 (ENSG00000184898) passed the integrated GWAS summary statistics with putative BE-causal cis-eQTLs by colocalization analysis with the threshold of PPH4 > 0.5 (Figure 6(b)). Figure 6(c) shows the SNP and SMR associations across the mQTL, eQTL, and BE GWAS at the RBM43 locus (Figure 6(c)). Figure 6(d)–(f) shows that the SNP signals associated with RBM43 were significant across the data from smoking GWAS, eQTL, and mQTL studies. The RBM43 expression level was negatively associated with BE outcome (beta SMR = −0.212). DNA methylation levels by probe cg00935895 showed a positive effect on BE (beta SMR = 0.267) and a negative effect on RBM43 expression (beta SMR = −0.303). Our results also suggest a putative mechanism wherein a lower DNA methylation level in the enhancer region of RBM43 upregulates the expression of RBM43 and subsequently decreases the BE risk (Figure 6(d)–(f)).

RBM43 gene screening, colocalization analysis, and three-step SMR analysis prioritized putative causal RBM43 gene with BE. (a) Intersection gene screening from SMR results for smoking and BE; (b) locus comparisons between cis-eQTLs and BE GWAS by colocalization analysis (PPH4 > 0.5); (c) locus plots illustrating consistent genetic effects from BE GWAS, cis-mQTL, and cis-eQTL near RBM43 (all minimum p < 1.0E-05). (d–f) Three-step SMR indicates significant causal relationships between RBM43 gene expressions and BE onset mediated by methylation (all HEIDI test p > 0.05). The r2 value indicates the LD between the variants and the top SNPs. BE, Barrett’s esophagus; eQTL, expression quantitative trait locus; HEIDI, Heterogeneity in Dependent Instruments; mQTL, methylation quantitative trait locus; SMR, summary data-based Mendelian randomization; SNP, single-nucleotide polymorphism.
Discussion
In this study, we first conducted MR analysis to explore the potential association between smoking and BE. Subsequently, we integrated the GWAS and cis-QTL data to identify putative genes that showed a pleiotropic/potentially causal association with smoking dependence and BE.
We confirmed that smoking dependence significantly increased the risk of BE and further identified significant genes that may be involved in the pathogenesis of both smoking dependence and BE. Our findings provide important insight into the mechanisms underlying the association between smoking dependence and BE. This study also identified potential therapeutic targets for effective prevention and treatment of BE.
BE is the precursor of EAC 25 and guidelines suggest screening for BE in populations with multiple risk factors. 26 Understanding the potential association between smoking and BE, as revealed by our MR study, may facilitate early intervention strategies, thereby reducing the likelihood of recurrence in populations with dependence on smoking. However, failure to screen high-risk individuals represents a missed opportunity for EAC prevention and early detection. 27
Our MR study provides strong evidence supporting a causal link between heavy cigarette dependence and increased risk of BE. Furthermore, multivariable MR analysis demonstrated a significant association between the number of cigarettes smoked per day and current tobacco smoking status and the risk of BE in comparison with other forms of smoking exposure, such as secondhand smoke. These findings are consistent with those of previous observational studies, which have indicated a potential relationship between smoking and BE.5,28 However, unlike observational studies, MR designs reduce the risk of confounding and reverse causation, thereby strengthening causal inference. 29 This robust methodology suggests that the observed associations are likely causal rather than merely correlational. 30 The consistency of these results with those obtained from multivariable MR analysis reinforces the reliability of our findings and underscores the importance of genetic factors in modulating the risk of BE among individuals with a propensity for heavy smoking dependence. Together, these analyses suggest that genetic variants that influence smoking behavior also play a role in the pathogenesis of BE.
Additionally, our bidirectional MR analysis was designed to assess the possibility of reciprocal causation between BE and cigarette smoking. These results support the hypothesis that cigarette smoking increases the risk of BE, with evidence in the reverse direction. The results were consistent with both previous MR studies 31 and observational studies 32 ; notably, our study also revealed a significant association in either direction. This could imply one of several scenarios: (1) both smoking and BE might influence each other over time. In this case, smoking could increase the risk of BE; however, the presence of BE might also influence smoking behavior, perhaps due to symptom exacerbation leading to changes in smoking habits. (2) There may be common genetic variants that influence both smoking behaviors and the risk of BE independently. This could mean that individuals who are genetically predisposed to smoking might also be predisposed to developing BE, but not necessarily because one directly causes the other; (3) there could be unmeasured confounders that affect both smoking and BE, such as socioeconomic status, dietary habits, or other lifestyle factors that were not accounted for in the analysis; and (4) it is also possible that the bidirectional analysis picked up indirect effects or pleiotropy, where genetic instruments affecting one phenotype also affect another phenotype through unrelated pathways. In our bidirectional MR analysis, the presence of heterogeneity indicates the influence of potential confounders. However, the analysis passed the test for horizontal pleiotropy, further confirming the potential genetic association between smoking and BE.
Our study employed a SMR approach to integrate a comprehensive set of meta-analytic cis-QTL information derived from the GTEx project.33,34 Integrative analysis identified the specific gene RBM43 as the top hit, exhibiting a negative association with both smoking dependence and BE. This protein functions as a tumor suppressor by regulating CCNB1 expression, thereby controlling the cell cycle. 35 Recent studies have revealed that RBM43 inhibits the expression of mitochondrial and thermogenic genes in a PGC1α-dependent manner, and the loss of RBM43 protects cells from cytokine-induced mitochondrial dysfunction. 36 Therefore, the regulatory mechanisms of RBM43 in smoking dependence and BE require further investigation. This discovery also highlights the potential role of the genetic regulation of gene expression in mediating the effects of smoking dependence on BE risk.
The observed bidirectional association between smoking and BE, where both forward and reverse MR analyses suggest a promoting effect of one on the other, presents intriguing findings but is not without limitations. One key limitation is the potential for pleiotropy or horizontal pleiotropy, where genetic variants influence multiple traits independently of the BE smoking pathway, which can lead to biased estimates in this MR study. Although the MR-Egger intercept analysis did not reach statistical significance (p > 0.05), suggesting no substantial directional pleiotropy, the significant Cochran’s Q test in the reverse analysis indicated heterogeneity among the instrumental variables, raising concerns regarding the validity of the genetic instruments used. This heterogeneity may reflect the presence of unaccounted confounders or nonlinear relationships that were not captured by the linear models typically employed in this analysis. Additionally, the bidirectional causality suggested by the analyses might be an artifact of shared genetic predispositions or lifestyle factors that influence both smoking behavior and the risk of developing BE, rather than a true causal relationship.
Additionally, the generalizability of our findings may be limited by the predominant European ancestry of the GTEx cohort, which requires replication in diverse populations. It is important to note that our study predominantly utilized data from European cohorts, limiting the applicability of our findings to diverse populations. Genetic architecture can vary significantly across ethnicities, potentially leading to distinct causal pathways and effect sizes. Although our results provide robust insights into the genetic mechanisms underlying smoking and BE in European populations, they should be interpreted with caution when applied to other ethnic groups. Future studies should aim to replicate our findings in diverse populations, including individuals of African, Asian, and Latin American descent, to ensure broader applicability and enhance the generalizability of our conclusions. Incorporating multiethnic datasets will help identify population-specific genetic effects and improve the precision of the analyses. Therefore, future studies should focus on functional validation of the identified genetic variants and their interactions with environmental exposure to better understand the complex etiology of BE.
The discovery of RBM43 opens new avenues for translational research aimed at developing novel therapeutic strategies for BE. Understanding how genetic variations influence susceptibility to BE can guide the development of personalized medical approaches that target underlying molecular mechanisms. 37 Such efforts could lead to the identification of new biomarkers for the early detection and more effective treatment of BE, potentially improving patient outcomes and reducing the burden of the disease on the healthcare system.
Conclusions
Our study provides compelling evidence of a causal relationship between heavy cigarette dependence and the risk of developing BE. Integration of cis-QTL data from GTEx has identified RBM43 as a key gene in this relationship, offering new targets for future research and potential therapeutic interventions. Public health strategies aimed at reducing smoking rates may be pivotal in lowering the incidence of BE and its subsequent complications. Further research is needed to confirm these findings and to explore the underlying mechanisms responsible for the observed associations.
Supplemental Material
sj-xls-1-smo-10.1177_20503121251316595 – Supplemental material for A bidirectional Mendelian randomization study integrating genome-wide association studies, expression quantitative trait locus, and methylation quantitative trait locus data revealed causal relationship between heavy cigarette dependence and Barrett’s esophagus
Supplemental material, sj-xls-1-smo-10.1177_20503121251316595 for A bidirectional Mendelian randomization study integrating genome-wide association studies, expression quantitative trait locus, and methylation quantitative trait locus data revealed causal relationship between heavy cigarette dependence and Barrett’s esophagus by Zhou An, Meichun Zeng and Xianhua Wang in SAGE Open Medicine
Footnotes
Acknowledgements
We express our gratitude to OpenGWAS (https://gwas.mrcieu.ac.uk) and the UK Biobank (![]() ) for providing the publicly available data for our study.
) for providing the publicly available data for our study.
Author contributions
All authors have made substantial contributions to the development and completion of this study. Z.A. was involved in the conception and design of the study; the acquisition, analysis, and interpretation of data; and drafted the article. M.Z. contributed to data analysis and revised the article. X.W. contributed to study design, data analysis, and article drafting. All authors provided final approval and agreed to be accountable for all aspects of the study.
Availability of data and materials
All data generated or analyzed during this study were included in the datasets of the OpenGWAS database (https://gwas.mrcieu.ac.uk), the UK Biobank (https://www.ukbiobank.ac.uk/), and the study’s ![]() .
.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
For the current research, it was not necessary to seek ethical approval since the initial investigation had already secured appropriate permission.
Informed consent
Informed consent was not required for this study because the original research participants provided written informed consent.
Consent for publication
Not applicable.
Trial registration
Not applicable.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.