
Abstract
Background:
Impact of drug wastage is a legitimate and persistent concern. Financial impact of drug waste is borne by the hospital network, patients, and healthcare systems. Measures to reduce drug wastage may have a positive impact throughout healthcare systems.
Objective:
This study investigated the stability and sterility of single-dose vials when repeatedly accessed with a closed system transfer device. By evaluating the sterility and stability, these results may be used to validate the extension of vial usage and lead to potential drug wastage reduction.
Methods:
Sterility testing was performed in accordance with US Pharmacopeia 71. A closed system transfer device was incorporated into simulated compounding tasks, utilizing growth media. Simulated compounding tasks were performed in the clinical environment, followed by incubation to stimulate growth. Stability testing was performed in accordance with US Pharmacopeia monographs at multiple timepoints post access. Test samples were comparatively tested via high-performance liquid chromatography to freshly opened vials at each timepoint.
Results:
No growth was observed in test samples. Control vials displayed growth, where appropriate. The drugs retained stability, when compared to freshly opened vials at 0, 24, 48, and 72 h, post access.
Conclusions:
This study confirms that closed system transfer devices do not contribute to microbial contamination of drug vials, following the repeated access, for up to 7 days and the tested drugs retained equivalent chemical stability for up to 72 h post access. This study may offer a manner by which a facility may assess single-dose vials’ sterility and stability, following repeated access by a closed system transfer device.
Background
Publications have shown that the financial impact of cancer drug wastage is substantial and increasingly borne by the patient.1–4 In the United States alone, this wasted drug material may exceed 3 billion USD in unused oncology drugs per annum. 5 Increased financial impact of cancer drug waste, which generally occurs for higher expenditure drugs, can manifest complications in fulfilling patients’ requirements in additional ways such as drugs shortages. Invariably, these costs are borne by the hospital network, patients, and healthcare systems.
Cost containment associated with drug expenditure in oncology is a well-known concern. Institutions worldwide are focused on ways to increase profit by decreasing drug expenditure. A recently published retrospective analysis of spending showed that total expenditure on antineoplastic agents in the United States rose from $26.8 billion in 2011 to $42.1 billion in 2016, and this number is expected to continue to increase with the continuous development of new targeted therapies. 6
In practice, pharmacies operate under the guidance of US Pharmacopeia (USP), and the majority of US states’ laws require pharmacies to comply with USP standards. The standards for sterile products are found in USP Chapter <797> which states that single-dose vials (SDVs) must be discarded if not used within 12 h after being opened when exposed to ISO Class 5 or cleaner air. 7 The practice of limiting the time of use for such vials to such a narrow window can contribute to the wastage of medications that would otherwise likely be used. This in turn directly affects drug expenditure, increasing the amount of purchases an institution must make.
While wastage is a definite concern in today’s healthcare landscape, the common concern of drug shortages can often be more clinically concerning. Oncology drug shortages have unfortunately become an ordinary part of practice and may have unintended consequences: delays in starting or continuing therapy, substitutions of less effective or potentially more toxic regimes, patients switching physicians or location, and disruptions or changes to clinical trials. 8 It is reasonable to conclude that shortages may be further exacerbated by the requirement to dispose of SDVs that may not be completely used following the 12-h post-access expiration requirement. The potential to use these drugs rather than throwing them out is apparent, and if proven safe, could reasonably be an effective mitigation tool for navigating the uncertain world of drug shortages.
The use of closed system transfer devices (CSTDs) is a key strategy in complying with USP Chapter <800> standards and minimizing risk to employees. The use of CSTDs is currently recommended for compounding of hazardous drugs and must be used when administering antineoplastic hazardous drugs, when the dosage form allows. 9 The use of CSTDs during medication compounding has become a widely accepted practice. 10 In addition to providing an additional level of safety to the user, evidence has shown11,12 that CSTDs can help to prevent microbial contamination when used with medications manufactured in drug vials.
Objective
The objective of this study was to investigate the outcomes on stability and sterility when using the ChemoLock™ CSTD (ICU Medical, San Clemente, CA, USA) and to determine if its use could potentially extend the time medications in SDVs could safely be used. By ensuring the sterility by microbial inspection and comparatively evaluating the stability against freshly opened vials of identical drugs, this may be used to validate the extension of SDVs beyond use date.
Methods
Sterility testing method
Performance of the compounding tasks to perform the sterility testing of the SDVs were done in a class II laminar fume hood (LFH). Laboratory personnel were trained in the use of the CSTD by the manufacturer and utilized training material that included product directions for use (DFU), in-service posters, and supplementary training video(s) for the CSTD. The performance of the sterility test method began and was completed in March 2020, at a cGMP analytical laboratory (Dynalabs LLC, St. Louis, MO, USA) registered with the US Food and Drug Administration (FDA), DEA, and Bureau of Narcotics and Dangerous Drugs licensed, as well as ISO 17025 accredited.
The primary, experimental laboratory standard test method utilized for the study was in accordance with USP 71, 13 sterility test. This standard is identified as to be used when sterility testing is required per USP 797 to extend beyond use dating.
To ensure the possibility of growth of gram positive, gram negative, anaerobic microorganisms, yeast, and mold, two types of growth media were utilized: tryptic soy broth (TSB) and fluid thioglycollate medium (FTM). Also, two temperature ranges (20°C–25°C and 30°C–35°C) were utilized during incubation to ensure optimum growth conditions for different microbes. This test method has been determined to be scientifically sound and robust. 9
Simulated compounding tasks were performed, including standard disinfecting of the septum, by utilizing both growth media, and transferring growth media to the sample vials to maximize the opportunity for contaminate growth.
Media vials of TSB and FTM were segregated to act as positive and negative controls; two for negative and three for positive controls. The vials were included in the compounding process by the placement of the control vials inside the LFH during the process, but not manipulated. These controls were then stored under identical conditions as the sample vials tested. Prior to incubation, each positive FTM control vial was inoculated with a maximum of 102 CFU (Colony Forming Unit) of one each of the following American Type Culture Collection (ATCC) strains: Clostridium sporogenes, Pseudomonas aeruginosa, and Staphylococcus aureus and incubated at 30°C–35°C for 5 days. Also prior to incubation, each positive TSB control vial was inoculated with a maximum of 102 CFU of one each of the following ATCC strains: Bacillus subtilis, Aspergillus brasiliensis, and Candida albicans and incubated at 20°C–25°C for 5 days.
Ten sample vials and five control vials were used for each growth media combination. On the day of the simulated compounding tasks, each vial was labeled with a preformatted label. Each vial septum was disinfected with sterile isopropyl alcohol IPA (isopropyl alcohol) 70% for 3 s and allowed to dry. Firstly, the CSTD Vial Adaptor was removed and inserted into the sample vial. Per the DFU, the CSTD vial septum was then disinfected by swabbing with sterile isopropyl alcohol 70% and the CSTD syringe connector attached to a syringe. Following the connection, 1 mL aliquot was drawn from each sample vial and stored at room temperature. The microbial turbidity growth of each of these sample vials was inspected at 0, 24, 48, and 168 h post preparation.
At the end of 168 h post preparation, each vial was provided to the microbiology laboratory under temperature control. Prior to the incubation at the respective temperature control, the microbiology technician ensured that the media solution was in contact with the vial surfaces. Each sample vial and negative control was incubated at the microbiology laboratory for 7 days at 20°C–25°C for and then an additional incubation at 30°C–35°C for 7 days. Vials were visually inspected for growth every 2 days and at the conclusion of the incubation period; the method of analysis was performed in accordance with USP 71.
Stability testing method
Preparation, compounding, and analysis tasks were performed at a NJ-DEP (Department of Environmental Protection) Certified, DEA licensed, and FDA registered analytical chemistry laboratory (ARL-EuTech, Highland Park, NJ, USA) following the approved protocol, and GMP/GLP guidelines. Performance of the primary, experimental laboratory standard test protocol was performed by analysts, previously trained on the usage of the CSTD system and began and completed in January 2020. All manipulation of test samples was performed inside a biological safety cabinet (BSC).
To track the stability of the SDVs, the analytical chemistry laboratory analyzed the four targeted compounds at 0, 24, 48, and 72 h post initial vial access using CSTD Vial Adaptor, and a syringe fitted with an appropriate CSTD syringe adaptor. The CSTD device was used for each “Test” vial and was analyzed alongside a fresh “Control” vial at each time interval. Each sample was analyzed in triplicate at the specified time interval by high-performance liquid chromatography (HPLC). Control vials were also included in the test method for each timepoint and analyzed drug combination to be included as reference standards. These control vials were freshly opened vials of identical drug medications. This process was repeated in triplicate for each test and control group.
Each SDV group was separated into the control group (SDV preparation without CSTD) and test group (SDV preparation with CSTD). Table 1, below, summarizes the testing protocol for each SDV group.
Testing protocol layout for each single-dose drug vial group.

The test vials were uncapped and the CSTD removed from packaging, inside the BSC, wiped with IPA swabs for 3 s and allowed to completely dry. The CSTD Vial Adapter was attached to the drug vial and the port swabbed with IPA and allowed to dry, per the device DFU. Following this, the test vials were accessed with the CSTD syringe, and a predetermined amount of drug was drawn from the vial. These steps were repeated for each SDV drug test group and for the control group without the use of a CSTD. Each test group vial and corresponding control group vials were sampled at 0, 24, 48, 72 h post access.
Sample preparation
Preparation of the samples to be used for the stability analysis was performed in accordance with USP monographs, as specified below:
Cytarabine
One milliliter sample was accurately weighed and transferred into a 100-mL volumetric flask and diluted to volume with water to prepare 1 mg/mL solution. Five milliliter of the 1 mg/mL solution was accurately weighed and transferred into a 50-mL volumetric flask and diluted to volume with water to prepare 0.1 mg/mL solution.
Fluorouracil
One milliliter sample was accurately weighed and transferred into a 100-mL volumetric flask and diluted to volume with water to prepare 0.5 mg/mL solution. One milliliter of the 0.5 mg/mL solution was accurately weighed and transferred into a 50-mL volumetric flask and diluted to volume with water to prepare 0.01 mg/mL solution.
Gemcitabine
One milliliter sample was accurately weighed and transferred into a 100-mL volumetric flask and diluted to volume with water to prepare 1.0 mg/mL solution. Five milliliter of the 1.0 mg/mL solution was accurately weighed and transferred into a 50-mL volumetric flask and diluted to volume with water to prepare 0.1 mg/mL solution.
Ifosfamide
Three milliliter sample was accurately weighed and transferred into a 250-mL volumetric flask and diluted to volume with water to prepare 0.6 mg/mL solution.
Each time samples were assayed according to the validated method described in the laboratory report. 14 Test methods were developed using corresponding USP standards as reference (conditions below).
The results of the drugs used as the test target compounds for this study of stability of the SDVs may be used as representative results, transferable to drugs that pose a financial burden through drug waste. These drugs were: cytarabine (100 mg/mL, NDC71288-109-20), fluorouracil (50 mg/mL, NDC16729-276-76), gemcitabine (100 mg/mL, NDC16729-419-03), and ifosfamide (50 mg/mL, NDC0143-9531001) and were sourced by ICU Medical and diluted with water to the sample concentrations indicated. Dibasic sodium phosphate, monobasic potassium phosphate, monobasic sodium phosphate, phosphoric acid, potassium hydroxide were obtained from Sigma-Aldrich (St. Louis, MO, USA). HPLC grade acetonitrile and methanol were obtained from GJ Chemical (Newark, NJ, USA). Water was generated in house. HPLC columns were generously donated by Microsolv Technology Corporation (Leland, NC, USA).
Analyses were performed on an Agilent 1100 HPLC system (Agilent Technologies, Santa Clara, CA) equipped with DAD. Data was acquired with ChemStation software (Chemstations, Houston, TX).
Results
Sterility
A total of 10 TSB vials and 10 FTM vials with the ChemoLock CSTD Vial Adaptor and CSTD syringe assembly were collected during different time points for up to 7 days (168 h) study period. No evidence of microbial contamination was noted in any test sample as shown in Table 2, below. This data indicates that the CSTD, when used in accordance with the product’s DFU and standard vial disinfecting processes, does not contribute to microbial contamination for up to 7 days. Controls were incorporated into the testing, as mentioned in the methods section. The positive controls displayed visual growth during the incubation period timeframes. The negative controls did not display growth.
Sterility data.

Stability
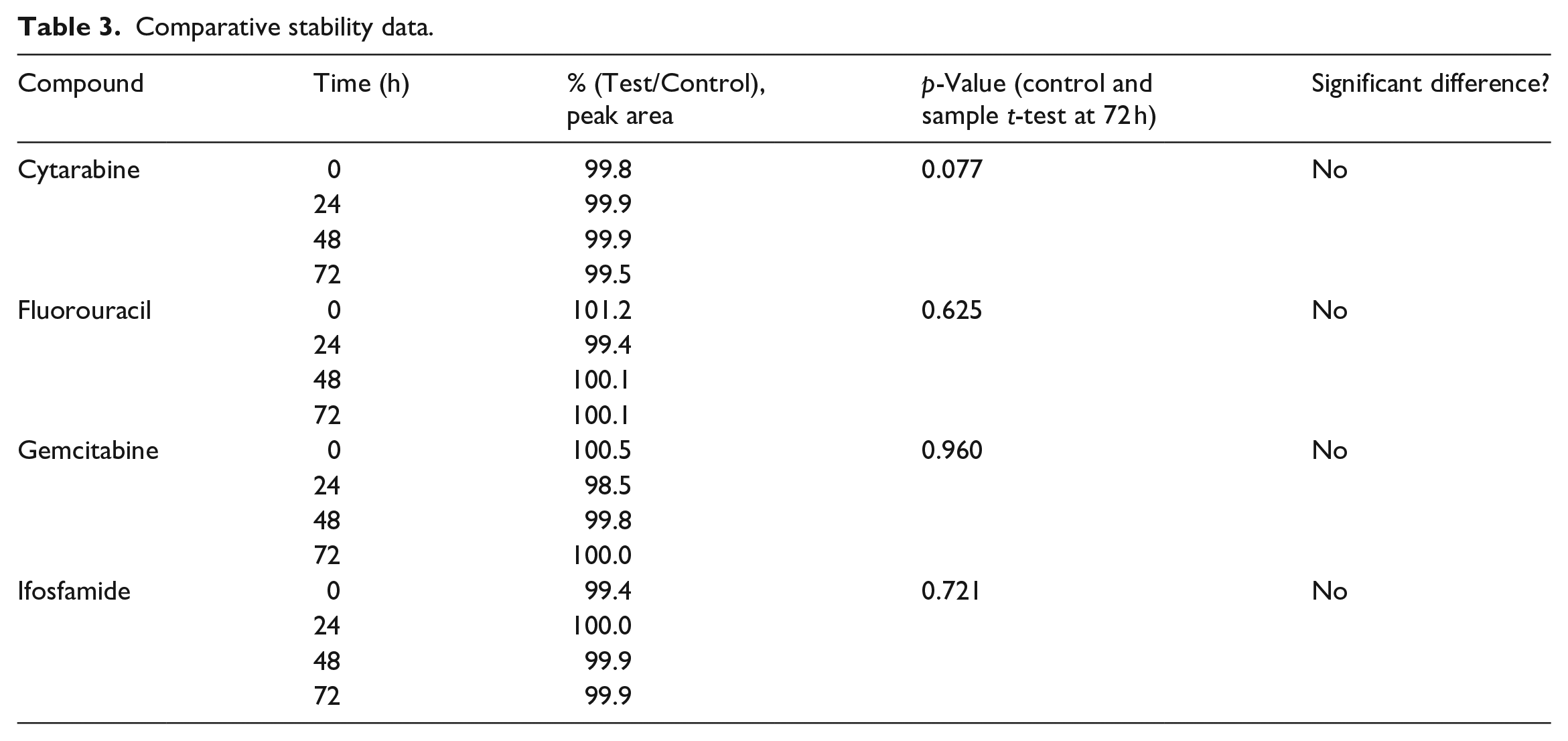
A total of eight test vials with four targeted compounds were analyzed at 0, 24, 48, and 72 h with a CSTD Vial Adaptor for the stability study period in triplicate. Control vials consisted of freshly prepared unopened vials with certificates of analysis indicating that the drugs were within the USP monograph limits (cytarabine—98%–102%, gemcitabine hydrochloride—97.5%–101.5%, fluorouracil injection—90%–110%, ifosfamide—98%–102%). These controls vial comparisons replicated conformity to USP monograms.
The HPLC chromatograms of the drugs and controls tested were (A) Cytarabine, 0.1 mg/mL in water. Column: Microsolv Cogent, RP C18, 5 µm, 100 A, 250 × 4.6 mm2, C/N:68518-25P; Mobile phase: Methanol: 0.73 g/L monobasic sodium phosphate and 1.4 g/L dibasic sodium phosphate in water (5:95); Flow rate: 1 mL/min; Injection volume: 10 µL; Detection: 254 nm; (B) Fluorouracil, 0.01 mg/mL in water. Column: Microsolv Cogent, RP C18, 5 µm, 100 A, 250 × 4.6 mm2, C/N:68518-25P; Mobile phase: Methanol : 0.73 g/L monobasic sodium phosphate and 1.4 g/L dibasic sodium phosphate in water (5:95); Flow rate: 1 mL/min; Injection volume: 20 µL; Detection: 254 nm; (C) Gemcitabine, 0.1 mg/mL in water. Column: Microsolv Cogent, RP C8, 5 µm, 100 A, 250 × 4.6 mm2, C/N:68508-25P; Mobile phase: 13.8 g of monobasic sodium phosphate and 2.5 mL of phosphoric acid in 1 L of water; Flow rate: 1.2 mL/min; Injection volume: 20 µL; Detection: 275 nm; (D) Ifosfamide, 0.6 mg/mL in water. Column: Microsolv Cogent, RP C18, 5 µm, 100 A, 300 × 3.9mm2, C/N: 68518-30P; Mobile phase: Water: Acetonitrile (70:30); Flow rate: 1.0 mL/min; Injection volume: 25 µL; Detection: 195 nm.
The results of the HPLC analysis are outlined in Table 3 and the actual chromatograms of test samples are shown in Figure 1, below.
Comparative stability data.

HPLC chromatograms of drugs tested.
For each compound, the independent two-sample t-test was performed between the Control and Test sample groups at 72 h to determine if there was any statistically significant difference between them as outlined in Table 3. Also included are figures demonstrating the peak area differences at 72 h for each compounded sample.
The test vials displayed similar mean concentration in comparison to the freshly opened vials. Comparative t-test indicate no significant statistically significant differences between the test and freshly opened vials.
Discussion
The results show that there is a potential cost-saving benefit of using a CSTD in pharmacy practice. Sterility testing show that the test articles, which included the CSTD, maintained the sterility of the system for up to 7 days when the vial remained capped with the CSTD Vial Adaptor and accessed only using a compatible syringe fitted with a CSTD and retained prolonged sterility, in comparison to USP standard use time, of up to 7 days after opening each vial. The stability results demonstrated no clinically significant reduction in stability for up to 3 days, after multiple accesses, in any of the four drugs that were studied as part of this work.
The results of the sterility testing, outlined in Table 2, indicate that there was no growth of any of the organisms in the sample vials after the initial and extended incubation period at the microbiological laboratory. These results were consistent with the negative controls. The positive controls showed growth, as expected. Sterility results from this testing confirm that even after repeated access with a CSTD, the microbiological sterility was not compromised.
Sterility testing has been evaluated in SDV accessed with a CSTD previously. 15 These studies highlight the potential cost-savings advantages of utilization of a CSTD to validate the extension of the beyond use date for SDVs.16,17 This study reinforces the previously published work but provides a reproducible robust method of evaluation that additional facilities may utilize to validate the supported extension. Individual facilities must evaluate these procedures within their own facilities to properly address any potential factors arising as a result of their own facilities’ environment, work practice, or workflow. Facilities should also consider specific regional regulation or guidelines and manufacturers’ instructions for use when evaluating the extension of any use time periods.
The results of the stability testing, as outlined in Table 3, show that there was equivalence in the test vials in comparison to freshly opened vials of the identical drug via HPLC chromatogram. The p values in Table 3 indicated that there was no significant difference in the results of this testing. The results of this testing show that even after 72 h, the SDV accessed with a CSTD was able to retain the drug stability.
Although the previously referenced publications clearly identify that sterility may be maintained, in both growth media and utilizing antineoplastic drug test vials, this study is the first, to date, that evaluates both the maintenance of the stability and sterility of the medication in SDVs, accessed repeatedly by CSTDs. If beyond use of the drug is to be extended, this must be based upon all pertinent factors, including the stability of the drug. If the drug that is to be administered to the patient is not proven to have retained the same chemical properties, within acceptable limits, as the time of manufacture, the efficacy of the administration and safety are potentially at risk. To show that the drug retains the same chemical properties and therefore the same efficacy, it should be compared by an accepted method to new or freshly opened vials, as performed in this study. Additionally, this study added a simple method by which a local compounding unit may also ensure that the commonly administered drugs in their facility may also retain their chemical stability profile. By applying this method to their own facility and medications, an individual facility may validate that their SDVs may retain stability when accessed with a CSTD, in comparison to freshly opened vials, or provide evidence to the contrary.
This data does not constitute as advice to alter or exceed established guidelines related to drug preparations. Any modification must comply with required steps to change the established guidelines, rules, and standards related to aseptic preparations and techniques, such as the procedural changes described in USP 797.
Limitations
The limitations of the study are the sample size, reliance on visual determination of contamination, and exclusion of initial contamination assessment. Determining contamination by visual inspection may be inferior to inoculating and incubating plates of growth media. Visual examination was chosen over plating to maintain a completely closed system, minimizing the possibility of an additional source of contamination, which might confound the results.
FDA and USP guidelines on use date for SDVs are based upon an assumption of contamination during initial access. Though none of the test samples demonstrated contamination, CSTDs cannot reduce initial puncture contamination risk.
Conclusion
We should highlight that the use of CSTD allows us to maintain the drug assay for up to 3 days which is a significant increase from the 12-h USP guideline. However, the current guidance from USP does not include the ability to extend the stability and/or sterility of SDVs beyond the aforementioned limitations. Therefore, additional work is required to establish the reliability of this data and to further improve the industry-wide guidance. This information may provide a template for an individual facility to evaluate their own processes and work environment to determine if extension of the beyond use date of certain drugs may be achievable.
It is reasonable to extrapolate that the use of this CSTD system could retain stability of other drugs, though similar testing of each drug is warranted and recommended. It is also reasonable to conclude that healthcare facilities could likely reduce drug expenditure if able to utilize SDVs for extended periods of time, as well as be able to navigate drug shortage situations more easily. Direction for future evaluations may include expansion of evaluation of drug stability to include other classes of drugs (i.e., antibiotics, biologics, etc.) or extension of time period beyond those tested in this evaluation.
Footnotes
Author contribution
JNH: writing—original draft, writing—review and editing. MW: methodology, investigation, writing—review and editing. RS: investigation, writing—review and editing. JB: writing—original draft, writing—review and editing, visualization.
Disclaimer
The purpose of this research is to demonstrate the potential use of the ChemoLock Closed System Device. Its publication does not constitute and must not be construed as promotion or advice to deviate from, alter or otherwise exceed the guidelines, rules and standard related to aseptic preparations and techniques, or in any way deviate, alter or exceed the use date or shelf life of the drugs. For any modification a user must comply with required steps and measures to change the established guidelines, rules, and standard related to aseptic preparations and techniques, such as the procedural changes described in USP 797.
Declaration of conflicting interest
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: JH has received honoraria for partner authorship on manuscript. MW and RS declare no relevant conflicts of interest or financial relationships. JB is an employee of ICU Medical.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding of the study was provided by ICU Medical.
Ethics approval
Ethics approval is not applicable for this study. This bench study was performed without the enrolment of human subjects, inclusion of human biospecimens, or inclusion of any identifiable private information or identifiable biospecimens.
Informed consent
Informed consent is not applicable for this study. This bench study was performed without the enrolment of human subjects, inclusion of human biospecimens, or inclusion of any identifiable private information or identifiable biospecimens.
Previous presentations
Study results presented as a poster at ASHP Midyear 2021.