
Abstract
Background:
Antibiotic exposure disrupts microbial communities in the gut and respiratory tract, causing functional changes that may have lasting health impacts and contribute to the spread of antibiotic resistance genes (ARGs) throughout life. However, age-stratified evidence of these effects, particularly in low- and middle-income countries (LMICs), remains limited.
Objective:
This systematic review assessed the impact of antibiotics on gut and respiratory microbiomes and resistomes in LMICs, with separate analyses for adults and children.
Data sources:
PubMed, Scopus, Web of Science, & ScienceDirect.
Methods:
A comprehensive literature search was conducted using a predefined search strategy and eligibility criteria to identify relevant studies from LMICs. Twenty-five studies met the inclusion criteria: 23 examined the gut microbiome, and 2 focused on the respiratory microbiome. Key outcomes included microbial diversity (alpha/beta/gamma), taxonomic shifts, resistome profiles, functional changes, and recovery potentials, stratified by age group and body site.
Results:
Antibiotic exposure was generally associated with reductions in microbial diversity and altered taxonomic composition, with children showing more pronounced and prolonged disruptions than adults. Analysis of resistome changes revealed a critical finding: while antibiotics consistently selected for ARGs matching the drug class administered, a substantial reservoir of non-matching, background ARGs, conferring resistance to beta-lactams, aminoglycosides, vancomycin, tetracyclines, was also highly prevalent across studies. This indicates a silent pre-existing resistome that is enriched by antibiotic pressure. ARGs were more abundant in adult resistomes, though functional changes occurred across age groups. Microbiome recovery was observed over time, but resistome recovery was limited.
Conclusion:
Antibiotic use significantly disturbs the gut and respiratory microbiomes and promotes ARG enrichment, especially in children, who demonstrate greater susceptibility and lower recovery potential. These findings emphasise the need for targeted antibiotic stewardship, improved microbiome recovery research, and enhanced resistome monitoring in LMICs.
Trail registration:
International Prospective Register of Systematic Reviews (PROSPERO), ID: CRD420250641394.
Keywords
Introduction
Antibiotics are among the most widely used and misused therapeutic agents in the world.1,2 Over the past two decades, their consumption has grown substantially, reflecting increased availability and persistent clinical need. In an extensive analysis of antibiotic use across 76 countries, Klein et al. 3 using the IQVIA MIDAS database reported that between 2000 and 2015, global antibiotic consumption increased by 65%, rising from 21.1 to 34.8 billion defined daily doses (DDDs). 3 This trend persisted in subsequent years, with a 16.3% (29.5–34.3 billion DDDs) increase recorded between 2016 and 2023. 4 Although the consumption rate decreased by 4.9% in high-income countries (HICs), it increased by 14.0% in low- and middle-income countries (LMICs). 4 While this trend reflects progress in access to essential medicines, it has also accelerated the spread of antimicrobial resistance (AMR), which now has escalated into a global crisis, with 4.95 million deaths associated with resistant infections in 2019 alone. 5 The highest burdens are borne by LMICs, where resistance emerges in parallel with expanding access, often in the absence of stewardship systems that might temper misuse or prevent the silent spread of resistant organisms.5,6
Alongside resistance, repeated exposure to antibiotics disturbs the human microbiome, which includes a diverse community of microorganisms that inhabit the human body, particularly those in the gut and respiratory tract.7,8 These communities are not passive bystanders. They shape immunity, influence nutrient absorption and guard against opportunistic infections. The gut microbiome, for instance, is crucial for nutrient absorption, mucosal immunity and colonisation resistance against enteric pathogens.9,10 The respiratory microbiome, though less densely populated, performs similar protective functions at the airway interface, influencing the outcome of infections and the host’s inflammatory response.11,12 Disruptions to these ecosystems can impair colonisation resistance, immune development, and metabolic balance.9,13 In early life, when the microbiome is still forming, these disruptions may steer microbial development onto entirely different trajectories and potentially lead to long-term health consequences. 14 In adults, cumulative exposure may erode resilience and embed resistance more deeply into the microbial landscape. 15
Although the relationship between antibiotic use and the human microbiome has been examined in several reviews and studies, the majority of this research has been conducted in high-income countries or analysed through global datasets that often obscure regional differences.16–19 These studies largely reflect patterns of antibiotic exposure, healthcare infrastructure, and microbial baselines that differ substantially from those found in LMICs. Recently, a systematic review assessed the impact of antibiotic use on the gut microbiome and resistome in children under 2 years in LMICs. 20 However, a broader assessment of the impact of antibiotics on the gut microbiome through the life stages of humans could not be done due to the study’s limitation on the age of study participants. In this systematic review, we extend the focus by synthesising data from LMICs on antibiotic-related changes in the gut microbiome with explicit attention to age-stratified outcomes and resistance profiles, as well as determining the recovery potentials of these profiles over time. Additionally, a novel synthesis of observations in the respiratory microbiome will be provided.
Method
Study design and systematic review protocol
This study was registered in the International Prospective Register of Systematic Reviews (PROSPERO) with registration ID: CRD420250641394 and followed the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) guidelines of 2020 21 (Supplemental Table 1).
Search strategy
We conducted a comprehensive literature search between 10 March 2025 and 24 March 2025 across several electronic databases, including PubMed, Scopus, Web of Science and ScienceDirect, to retrieve relevant articles, applying no restrictions on publication year or language. Full-text/open-access filters were applied across all databases, and the ‘Humans’ filter was additionally applied in PubMed. The search terms included keywords such as ‘Antibiotics’, ‘Gut microbiome’, ‘Gastrointestinal microbiome’, ‘Respiratory tract microbiome’, ‘Resistome’, and were designed to capture studies from all LMICs as classified by the World Bank (Supplemental Table 2). Furthermore, for databases that included a ‘country’ category, LMIC filters were applied. In addition, we performed supplementary searches by screening reference lists of included articles for relevant studies not indexed in the databases.
Study selection and eligibility criteria
The studies identified from individual databases were first exported and compiled in Zotero (Version 7.0.15), where they were merged into a single RIS file. This file was then imported into Rayyan, an online platform designed for systematic review screening, where duplicate studies were manually identified and resolved. The screening process was conducted in two stages. First, titles and abstracts were screened to assess their relevance based on the predefined inclusion and exclusion criteria. Second, the full texts of potentially eligible studies were retrieved and reviewed in detail. Duplicate removal and both stages of screening were conducted independently by two reviewers (S.N.-A.Y. and A.A.-D.). Disagreements were resolved through discussion, and unresolved cases were addressed by a third reviewer (E.S.D.).
Inclusion criteria
Studies were eligible for inclusion if they met all of the following conditions:
Conducted in LMICs as classified by the World Bank.
Investigated the impact of antibiotic use on the gut or respiratory microbiome and resistome, including both interventional studies (e.g. randomised controlled trials) and observational studies (e.g. cross-sectional, cohort, or case-control, whether prospective or retrospective).
Presented original research involving human subjects.
Exclusion criteria
Studies were excluded based on the following criteria:
Did not clearly specify antibiotic use or focused solely on non-antibiotic interventions.
Were abstracts, review articles, letters to the editor, case reports, research notes, addendums, systematic reviews or grey literature.
Conducted in high-income countries (HICs).
Did not provide stratified data by both age group (adults vs children) and setting (LMICs vs HICs), as required for the analysis.
Involved animal experimentation on gut or respiratory microbiome and resistome.
Data extraction
Data from the included studies were collected using a structured data extraction form that was designed in advance and tested on a small set of studies before full application. Two reviewers (S.N.-A.Y. and A.A.-D.) independently extracted the data, and any differences were first discussed; if agreement could not be reached, a third reviewer (E.S.D.) resolved them. The form covered key details such as author names, publication years, study years, study locations, microbiome types (gut and respiratory tract), study design, sample types (and collection timepoints), sample size, population characteristics (including age), antibiotics administered (dosage and duration), outcome measures (microbiome/resistome profiles, functional changes and recovery patterns), genomic methods, statistical methods, follow-up duration, health status and antibiotic history.
Specific attention was given to bacterial taxonomic composition (phylum, genus, species, family, order) and their ecological roles, particularly shifts post-antibiotic exposure. Because the included studies reported bacterial groups at different taxonomic levels, we recorded all levels provided; however, since most studies reported at the phylum level, this served as the common level for comparison. For studies that reported other classifications, the corresponding phylum was verified using the National Center for Biotechnology Information (NCBI) taxonomy. Abbreviations were written in full (e.g. E. coli as Escherichia coli), and spelling variations were corrected according to accepted nomenclature. To ensure accuracy, extracted data from both reviewers were cross-checked, and this quality control process helped minimise the risk of errors and improve reliability. The data were stratified by microbiome type (gut vs respiratory) and population subgroup (adults vs children) to enable targeted analysis.
All information was then organised in Microsoft Excel using predefined fields, which facilitated consistency, comparison across studies, and the identification of patterns related to antibiotic impacts on human microbiome and resistance.
Quality assessment
Three quality appraisal tools were employed in assessing the strength of individual studies. The Cochrane RoB 2 tool was employed in the assessment of randomised controlled trial studies. 22 The tool involves five domains, which are used to assess the quality of the studies with gradings pegged at low risk, some concerns, and high risk (Supplemental Table 3).
The National Heart, Lung, and Blood Institute (NHLBI) study quality assessment tool was used to assess the strength of observational cohort and cross-sectional studies. 23 There are 14 questions in which each answer was scored 0 or 1 based on whether the answer was ‘yes’ or ‘no’. N/A was used to indicate that the question was not relevant. Studies with scores of 10–14 were graded as good, indicating a low risk of bias; those with scores of 5–9 were fair, while those with scores ranging from 0 to 4 were graded as poor, indicating a high risk of bias (Supplemental Table 4).
The Joanna Briggs Institute (JBI) critical appraisal tool for case-control studies was used to assess the strength of case-control studies. 24 There are 10 questions for which each answer was scored 0 or 1 based on whether the answer was ‘yes’ or ‘no’. N/A was used to indicate that the question was not relevant. Studies with scores of 8–10 indicated a low risk of bias, those with scores of 4–7 indicated a moderate risk of bias, and those with scores less than 4 indicated a high risk of bias (Supplemental Table 5).
Result
Search and screening results
A total of 406 studies were retrieved from the initial search across four databases: 205 from PubMed, 143 from Scopus, 46 from Web of Science, and 15 from ScienceDirect. An additional 10 studies were identified through citation chaining, resulting in a total of 416 included studies (Figure 1). After 99 duplicate articles were deleted, titles and abstracts of 317 articles were screened based on eligibility criteria. A total of 69 studies were considered eligible and subjected to full-text evaluation. After full-text examination, 25 articles were eligible for inclusion, in which a total of 23 studies assessed the impact of antibiotics on gut microbiome, while 2 studies assessed the impact on respiratory microbiome.

PRISMA flowchart of study selection.
Overview and description of included studies
All included studies (n = 25) were published between 2017 and 2025, with most of them being conducted between 2015 and 2017. Most studies were conducted in China (n = 8), followed by Niger (n = 5), Burkina Faso (n = 4), India (n = 2). The remaining countries, Bangladesh, Uganda & Zimbabwe, South Africa, Botswana, Malawi and Cambodia had one study each. The majority of the studies included were randomised controlled trials (n = 13). This was followed by observational studies (n = 12) (including prospective cohort (n = 6), cross-sectional (n = 3), retrospective cohort (n = 1), case–control (n = 1) and prospective functional (n = 1) studies; Tables 1, 2 and 8).
Characteristics of adult gut microbiome and resistome studies.

↓, decrease; ↑, increase; ARGs, antibiotic resistance genes; HE, hepatic encephalopathy; N/A, not applicable; PCoA, principal coordinate analysis; RCT, randomised controlled trials; WGS, whole genome sequencing.
Characteristics of paediatric gut microbiome and resistome studies.

↓, decrease; ↑, increase; ARGs, antibiotic resistance genes; N/A, not applicable; OTU, Operational Taxonomic Units; PCoA, principal coordinate analysis; RCT, randomised controlled trials; WGS, whole genome sequencing; LC-MS, liquid chromatography-mass spectrometry.
A total of 23 studies focused on the gut microbiome, while 2 studies investigated the respiratory microbiome. Among the gut microbiome studies, 6 were exclusively conducted in adult populations (Table 1), and 17 were specific to children (Table 2). Both respiratory microbiome studies were child-focused (Table 8), reflecting the predominant research focus on paediatric populations, likely due to the higher clinical burden of respiratory infections and antibiotic use in early childhood. The study populations spanned a broad age range: adults were between 18 and 70 years old, while children ranged from ⩽7 days to 13 years of age. Gut microbiome studies utilised either faecal samples (15 studies) or rectal swabs (8 studies) for microbial analysis. In contrast, the respiratory microbiome studies employed bronchoalveolar lavage fluid from the lower respiratory tract and nasopharyngeal swabs from the upper airway in separate studies. All studies documented the collection timepoints of the samples. Regarding participant health status prior to antibiotic exposure, 20 studies provided this information. Of these, five reported healthy individuals, while the remaining studies involved participants with underlying health conditions (Tables 1, 2 and 8).
Of the 25 studies included, 24 reported the specific antibiotics or antibiotic classes used, either as therapeutic interventions, treatment regimens, or prophylactic agents (Tables 1, 2 and 8). One study did not specify the antibiotic involved in the intervention (Table 2). Across the studies, outcomes were generally compared with either placebo groups or untreated controls. All studies detailed the methods used to assess microbial diversity and composition as well as resistome changes. These included statistical and genomic methods. Eighteen studies provided information on participants’ prior exposure to antibiotics before the intervention. Among these, 8 studies involved antibiotic-naïve individuals, while 10 included participants who had been previously exposed to antibiotics. Notably, one study categorised as antibiotic-naïve involved mothers who had received antibiotics, while their neonates remained unexposed (Table 2).
Alpha diversity metrics were determined by the Shannon index (16 studies), followed by the Inverse Simpson index (10 studies). The Chao1 index and operational taxonomic unit count were used less frequently, with four and two studies, respectively (Tables 1 and 2). Beta diversity metrics were determined by the Bray-Curtis dissimilarity index (7 studies), followed by principal coordinate analysis (PCoA) and Euclidean distances (3 studies each), Jaccard distances, and the unweighted UniFrac algorithm (2 studies each). The less frequently used indices (one study each) were Shannon, Inverse Simpson and Morisita-Horn (Tables 1, 2 and 8). Gamma diversity metrics were determined by both the Shannon and Inverse Simpson indices (two studies each) (Table 2). Various genomic methods were employed across studies. Metagenomic sequencing was the most frequently used method (12 studies). This was followed by 16S rRNA gene sequencing (8 studies) and Shotgun metagenomic sequencing (4 studies). Whole genome sequencing (WGS), targeted metagenomic sequencing (mSWEEP), untargeted metabolomics by liquid chromatography-mass spectrometry (LC-MS), and resistance gene identifier were less frequently utilised (one study each) (Tables 1, 2 and 8).
For studies that did not assess or report on a specific category, ‘N/A’ was used to indicate that the category was not applicable.
Gut microecology profiles
Adult microbiome and resistome
Out of six studies, four analysed the alpha diversities of the microbiome, while five studies determined the beta diversities. Four studies examined bacterial taxa composition after antibiotic exposure and observed significant shifts in composition (Supplemental Table 7). The four studies observed a variety of bacterial species segregated according to taxonomical classifications (Supplemental Table 7). The most frequently reported taxonomic rank was the bacterial phylum. The notable phyla across all studies were Firmicutes, Proteobacteria, Actinobacteria and Bacteroidetes. Ecological roles played by these species across all studies include beneficial/commensal species, opportunistic pathogens, commensal species and commensal/opportunistic pathogens (Supplemental Table 7). Five studies analysed the resistome outcomes after exposure and observed prevalent antibiotic resistance genes (ARGs) (Table 1). Four studies specifically reported on the ARGs identified. These genes include: aminoglycosides (aph(2′)), sulphonamides, beta-lactams, carbapenems, chloramphenicol, macrolides, quinolones (qnrB7), tetracyclines (tetM), trimethoprim, streptomycin, and rifampicin (notably rifampin ADP-ribosyltransferase (arr)) resistance genes. Others include vancomycin (vanC, vanY-A, vanD, vanRG, vanRC), fosfomycin, phenolic compounds, and cephamycin resistance genes. Only a few studies (n = 2) reported on functional changes following antibiotic exposure. Five studies conducted longitudinal follow-ups to assess the recovery potential of the microbiome and resistome. Three studies reported full recovery after treatment, whereas two observed only partial recovery. In terms of resistome recovery, three recorded no recovery after follow-up, and two noted partial recovery (Table 1).
Child microbiome and resistome
Out of 17 studies, 14 reported on microbial diversity outcomes (alpha = 13, beta = 8, gamma = 3) (Table 2). Among these, 13 studies documented significant shifts in bacterial taxa composition post-exposure (Supplemental Table 8). The 13 studies observed a variety of bacterial species segregated according to taxonomical classifications. The most frequently reported taxonomic rank was the bacterial phylum. The notable phyla across all studies were Firmicutes, Proteobacteria, Actinobacteria and Bacteroidetes. Ecological roles played by these species across all studies include beneficial/commensal species, opportunistic pathogens, commensal species, pathogenic species, and commensal/opportunistic pathogens (Supplemental Table 8).
Eleven studies analysed the resistome outcomes after exposure and observed prevalent ARGs (Table 2). Detected genes include: aminoglycosides (aac(6′)-Ii), macrolides (ermQ, ermF, ermT, ermX and ermG), beta-lactams (blaCFXA3, blaCFXA, cepA and blaOXA), and trimethoprim (dfr) resistance genes. Other genes include nitroimidazoles, sulphonamides (sul), tetracyclines (tetO, tetQ, tetS, tetB(P), tet32 and tetX), and vancomycin (vanG and vanC). Additionally, two studies reported the presence of intrinsic resistance genes (mdtEFOP, acrDEF and emrKY) in Escherichia coli. A limited number of studies (n = 5) examined functional changes following antibiotic exposure. Four of these studies reported alterations in functional metabolic pathways, while one study additionally noted immune dysregulation alongside metabolic changes. Eleven studies conducted follow-ups to assess the recovery potential of the microbiome and resistome over time. Two of these studies reported full microbiome recovery post-treatment, two observed partial recovery, and another seven found no recovery. With regard to the resistome, three studies documented full recovery, while four studies observed no recovery (Table 2).
Comparative outcomes in adults and children
Generally, there was stable or reduced alpha diversity in adults as compared to a consistent reduced diversity in children as measured by the metrics. Beta diversity trends showed consistent reduction in adults as compared to stable or reduced diversity in children. There was an overall reduction in gamma diversity in children after antibiotic exposure while no data were reported for adults (Table 3). Bacterial taxa composition showed consistent significant changes across population groups (Supplemental Tables 7 and 8). In terms of specific taxa composition, phyla comparison showed that, Firmicutes (72.0% vs 28.0%), Proteobacteria (79.5% vs 20.5%), Actinobacteria (75% vs 25%), Bacteroidetes (66.7% vs 33.7%), and Verrucomicrobia (80.0% vs 20.0%) were most predominantly found in children gut microbiome as compared to adults. Candidatus Saccharibacteria (50.0% vs 50.0%) were equally found in both populations. The remaining phyla, Fusobacteria, Cyanobacteria, Desulfobacterota, Spirochaetes and Euryarchaeota were only found in child gut microbiomes (Figure 2).
The impact of antibiotic treatment on gut microbiome diversity in adults versus children.


Differential abundance of gut microbial phyla between age groups.
A general overview of the ecological roles played by these bacterial species was determined. It was observed that children had more beneficial/commensal (66.7% vs 33.3%), opportunistic pathogens (74.4% vs 28.6%), commensal (71.4% vs 28.6%), and pathogenic (100% vs 0.0%) bacterial species as compared to adults. There was a greater presence of commensal/opportunistic pathogens in adults (100% vs 0.0%) as compared to children (Figure 3). Both adults and children showed a general increase in ARGs following antibiotic exposure (Table 3).

Functional profile of the gut microbiome: adults versus children.
Antibiotics and antibiotic resistance genes
In adult microbiome studies, four major antibiotic classes were reported, with beta-lactams being the most predominant (66.7%) (Table 4 and Figure 4). These included subclasses such as cephalosporins, carbapenems and penicillin. The remaining classes were less frequently reported. In paediatric studies, five major antibiotic classes were identified. Macrolides were the most commonly used (48.1%), followed by beta-lactams (25.9%; cephalosporins and penicillin) and trimethoprim-sulfamethoxazole (18.5%), while other classes were reported less frequently (Table 4 and Figure 4).
Antibiotic prescription patterns by age group: adults versus children.


Proportional distribution of administered antibiotics by age group.
Overall, 16 ARGs were observed from all the gut microbiome studies (n = 23) combined with 14 being frequently observed in adult studies and 9 being observed in paediatric studies. Seven of these ARGs were consistently observed across both age groups (Supplemental Table 9). In terms of specific ARGs and frequency of occurrence, beta-lactam and vancomycin resistance genes were the most frequently reported in adults, followed by aminoglycosides and tetracyclines. Other ARG classes appeared less often (Supplemental Table 10). In children, macrolide resistance genes were the most predominant, followed by beta-lactam, tetracycline, trimethoprim, E. coli intrinsic, and sulphonamide resistance genes (Supplemental Table 10).
Among the 23 studies included, 14 provided sufficient data to examine linkages between administered antibiotics and ARG profiles (Table 5). Across both adults and children, antibiotics consistently selected for resistance genes corresponding to the administered antibiotic classes. In nearly half of these studies (6/14), additional non-matching or background ARGs were also detected, indicating collateral enrichment within the gut resistome, while the remaining eight reported only ARGs directly associated with the administered antibiotics.
Antibiotic-induced resistome: matching and non-matching ARG emergence in adults and children.
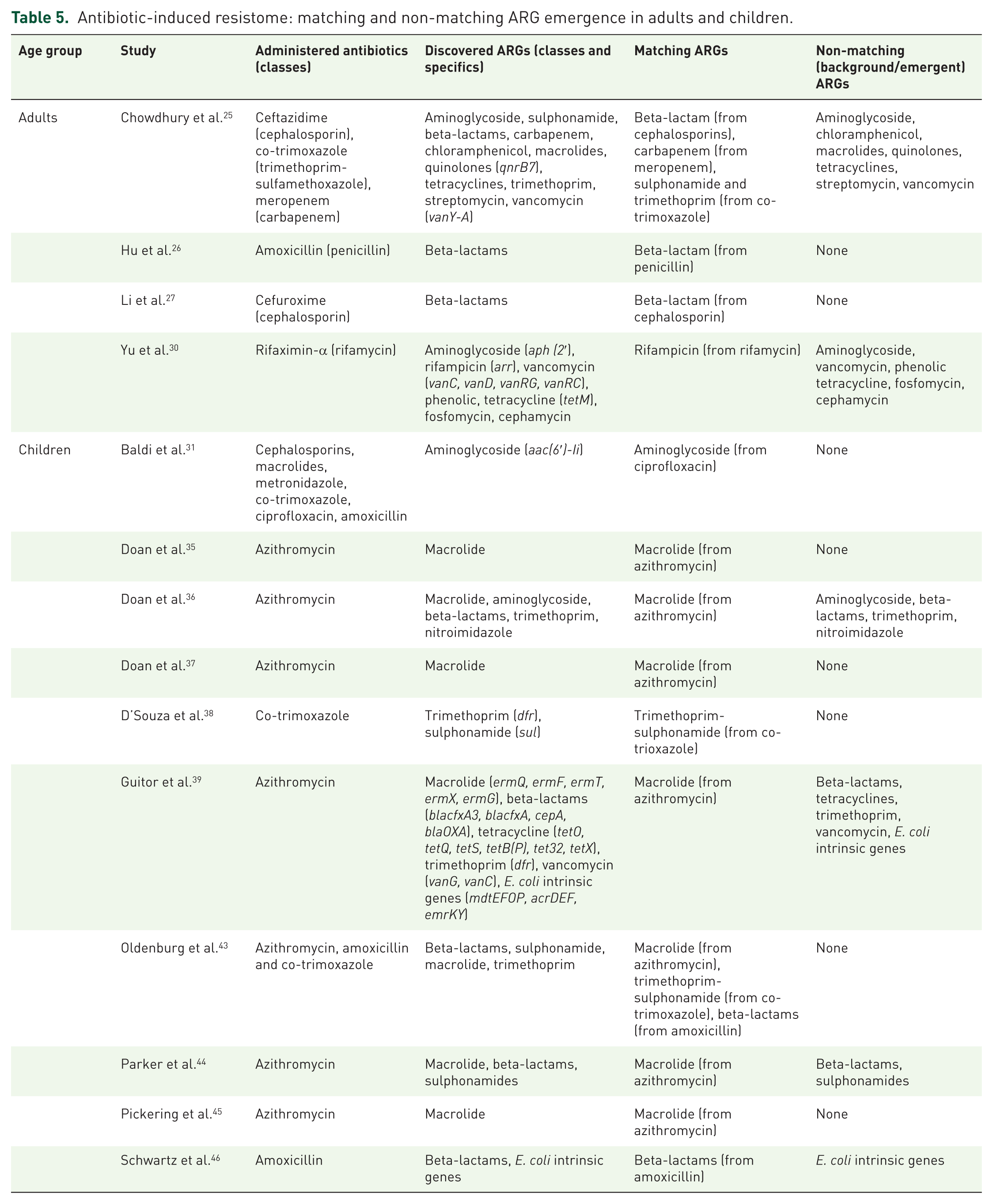
Health status, recovery, and functional outcomes
Participants were stratified from 18 of 23 studies into three health status categories: chronic illness, acute illness, and healthy (Supplemental Table 11). Across both adults and children, antibiotic exposure resulted in major reductions in microbiome diversity across all groups, accompanied by significant compositional shifts. Losses were most evident in alpha and beta diversity, with occasional gamma diversity reductions in children. These changes were consistently associated with increased ARG abundance, irrespective of age or health status (Table 6).
Gut microbiome and resistome variations by health status and age group.

ARGs, antibiotic resistance genes; ↓, decrease; ↑, increase.
In terms of recovery, evidence from 16 of the 23 studies indicated that adults generally demonstrated greater microbiome recovery than children following antibiotic exposure (Supplemental Figure 1). However, both age groups struggled with resistome recovery, though full restoration was occasionally observed in children (Supplemental Figure 2). Recovery patterns also varied by health status across 13 of the 23 studies (Table 7). In adults, chronic illness was associated with the most favourable microbiome recovery, often reaching full restoration, but resistome recovery remained only partial or absent. Acute illness in adults showed the poorest outcomes, with little to no recovery in either the microbiome or resistome, while healthy adults demonstrated mixed microbiome recovery and only partial resistome recovery. In children, microbiome recovery was generally limited, with most healthy and acutely ill populations showing no recovery, though one long-term study reported delayed restoration. Interestingly, children with chronic illness exhibited partial microbiome recovery alongside full resistome recovery, suggesting that ARG clearance can occur independently of microbial restoration. Overall, microbiome recovery was inconsistent, and resistome recovery was frequently incomplete or uncoupled from microbiome recovery patterns (Table 7).
Recovery potential of the gut microbiome and resistome stratified by participant health status.
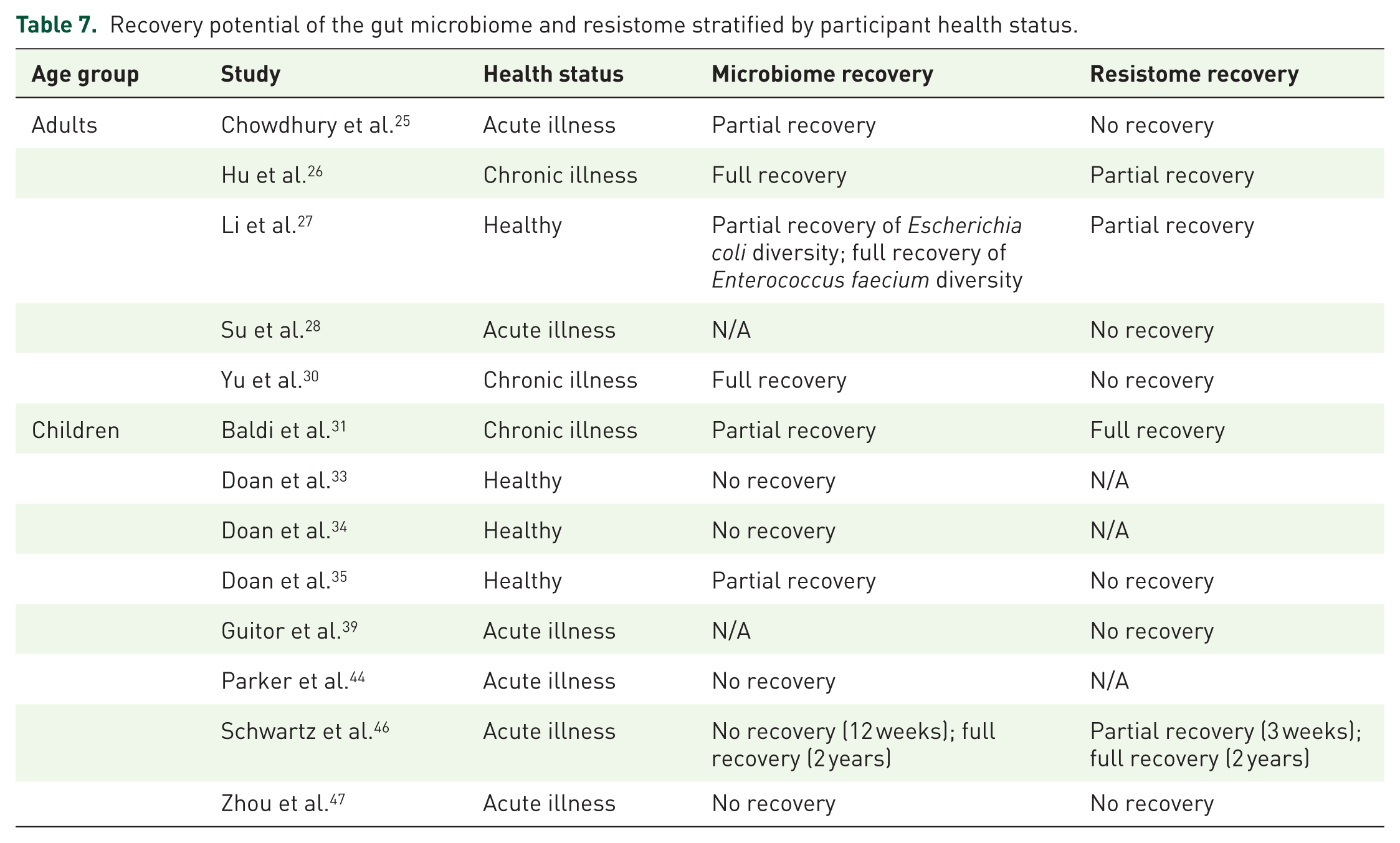
Among the seven studies that looked at functional outcomes, most reported disruptions to key metabolic processes in both adults and children, affecting pathways such as amino acid, cholesterol, and bile metabolism. In children, antibiotics were also linked to immune dysregulation, with reduced innate immune activity and loss of important gut metabolic functions. Some antibiotics further promoted increased antibiotic resistance-related pathways, while one adult study highlighted a serious clinical consequence: bloodstream infection driven by Klebsiella pneumoniae overgrowth (Supplemental Table 12).
Respiratory microecology profiles
Child microbiome and resistome
Overall, there was stable or reduced microbiome diversity and significant changes in bacterial taxa composition after antibiotic exposure in acutely ill participants (Table 8) (Supplemental Tables 13 and 14). In terms of specific taxa composition, Firmicutes (42.9%) were the most predominant phylum followed by Proteobacteria (35.7%). The rest of the phyla were reported less frequently (Supplemental Figure 3). In terms of ecological role, pathogenic bacteria (35.7%) were the most abundant (Supplemental Figure 4). Antibiotic resistance genes (ARGs) consistently increased across studies, with all reports detecting ARGs, including macrolides (macB), fluoroquinolones (patA-B), nitroimidazoles (msbA), peptides (bcrA), beta-lactams, and tetracyclines (tetT) (Table 8).
Characteristics of paediatric respiratory tract microbiome and resistome studies.

↓, decrease; ↑, increase; ARGs, antibiotic resistance genes; N/A, not applicable; NRMPP, non-refractory Mycoplasma pneumoniae pneumonia; RMPP, refractory Mycoplasma pneumoniae pneumonia; MALDITOF-MS, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry.
Regarding antibiotic exposure, four major classes were identified (Supplemental Table 15), with beta-lactams (37.5%) most frequently reported, followed by macrolides and fluoroquinolones (25% each) (Supplemental Figure 5). Only one study reported background ARGs not corresponding to administered antibiotics (Supplemental Table 16). Functional outcomes were rarely assessed; only one study documented altered functional metabolic pathways. Additionally, one study documented full microbiome recovery after follow-up. No data were available regarding resistome recovery (Table 8).
Discussion
Antibiotics are known to reshape the human microbiome, yet a comprehensive understanding of how these effects vary across the human lifespan remains unclear. While a previous systematic review by Luchen et al., 20 which focused on gut microbiome and resistome alterations in children under 2 years in LMICs, has provided valuable insights, their scope was limited by a narrow age range. Consequently, a broader, age-stratified assessment from early life through adulthood has been lacking. This systematic review addresses that gap by synthesising data from LMICs to explicitly examine antibiotic-related changes in the gut microbiome and resistome across different age groups, including adult populations, and to determine their recovery potential over time. Additionally, it incorporates evidence on antibiotic impacts on the respiratory microbiome and resistome, particularly in children, addressing an important but under-investigated aspect of this microecology.
Gut microbiome and resistome (adults vs children)
Microbial diversity and composition are fundamental aspects of the human microbiome, shaping the structure and function of microbial communities.50,51 These diversities are typically described using three interrelated metrics: alpha, beta, and gamma diversity. Alpha diversity reflects the richness (number of distinct taxa) and evenness (the relative abundance of those taxa) within an individual sample, offering insight into the internal complexity of a microbial community.52,53 Beta diversity, on the other hand, captures the variation in community composition between samples within the same habitat, emphasising differences in the identity and presence of taxa across samples.53,54 Gamma diversity represents the total richness observed across all samples within a given habitat, providing a measure of overall ecosystem-level diversity. 53 Together, these classifications offer a comprehensive framework for understanding microbial variation at both local and broader ecological scales.
The observed patterns of microbial diversity changes following antibiotic exposure as observed in this review, show the complex and age-dependent impact of these treatments on the gut microbiome. While adults often exhibit either stable or reduced alpha diversity, and children demonstrate a more consistent reduction across several diversity metrics, the overall trend points to a general loss of microbial diversity irrespective of age. These findings align with those of Konstantinidis et al., 55 who emphasised the detrimental impact of antibiotics on gut microbiome diversity in both adults and children. They noted that antibiotic administration, whether for therapeutic or prophylactic purposes, disrupts the microbial ecosystem by altering the abundance and composition of bacterial species. This disruption can lead to dysbiosis,56,57 which may alter the normal proportions and balance of beneficial bacteria and potentially promote the overgrowth of opportunistic or pathogenic species, thereby leading to potential adverse health conditions.
Antibiotic treatment was associated with distinct shifts in the predominant bacterial phyla within the gut microbiomes of both adults and children. The most commonly identified phyla across all participants included Firmicutes, Proteobacteria, Actinobacteria, Bacteroidetes and Verrucomicrobia. Notably, children exhibited a greater richness of bacterial phyla compared to adults, reflecting the broader taxonomic range and typical developing nature of the paediatric gut microbiome. 15 This broader range of bacteria included beneficial/commensals like Bifidobacterium alongside numerous other bacterial groups. In contrast, the adult microbiome demonstrated a more consolidated phylum-level profile, consistent with its greater maturity and ecological stability. These observations align with the established understanding that the developing gut ecosystem in children is more susceptible to colonisation by diverse taxa, while the adult microbiome tends to exhibit greater resilience to perturbation. 58
Furthermore, it is important to note that antibiotic administration resulted in varying effects on bacterial species. Some showed significant increases or decreases in abundance, while others remained relatively unchanged despite exposure. For instance, in one study involving participants diagnosed with severe acute diarrhoea, enteropathogenic Campylobacter species were detected in the microbiome. 39 Treatment with azithromycin led to a gradual reduction of these pathogens. This outcome was consistently observed across studies that identified Campylobacter and used azithromycin as the intervention.35,37,40,44 These findings are supported by the work of Luchen et al., 20 who also reported in their systematic review that azithromycin played a significant role in lowering the burden of enteric pathogens. Evidence by Guga et al. 59 and Rakita et al. 60 further elaborates on the effective potential of azithromycin. However, some notable enteropathogenic species like Shigella, Salmonella and Escherichia-Shigella remained prevalent even after antibiotic treatments,31,32,41 thereby portraying evidence of the current world issue on AMR.
This review observed that adults were frequently exposed to multiple, often potent classes of antibiotics primarily due to hospital-based or complex infection treatments. Most of these antibiotics belonged to the beta-lactam class, encompassing several subclasses such as penicillin, cephalosporins and carbapenems. These broad-spectrum agents are considered frontline treatments for severe bacterial infections but are also associated with strong selective pressure that promotes the emergence and spread of multidrug-resistant organisms. 61 Studies have shown that prolonged or repeated beta-lactam use in adults is linked to increased risks of resistant infections and disruption of the gut microbiome,62,63 leading to adverse long-term health effects including increased susceptibility to opportunistic infections (e.g. C. difficile infection)63,64 and altered immune responses. 63 In contrast, children were more frequently treated with targeted, first-line antibiotics such as azithromycin, a macrolide predominantly used for treating common enteric infections in outpatient settings. 65 However, extensive utilisation of this antibiotic has been observed to foster the proliferation of resistant strains capable of serving as reservoirs for resistance genes, 65 a finding well-substantiated throughout this review.
Taken together, these contrasting exposure patterns confirm that this difference in antibiotic treatment reflects age-specific prescribing practices and underlying infection profiles but also suggests differing impacts on microbial diversity and resistance gene carriage between adult and paediatric populations.
In terms of AMR, both adult and paediatric studies exhibited a notable presence of ARGs. Some of these common resistance genes include those conferring resistance to aminoglycosides, beta-lactams, macrolides, sulphonamides, tetracyclines, trimethoprim, and vancomycin. Interestingly, despite the limited number of studies focusing on adults, a greater number of distinct ARGs were observed in adult microbiomes compared to those of children (14 vs 9 resistance genes). This suggests that adult microbiomes may harbour a broader resistome, which could be associated with greater cumulative antibiotic exposure over time. This observation is understandable, as the likelihood of antibiotic exposure tends to increase with age. From infancy to adulthood, individuals often encounter various health conditions that necessitate repeated antibiotic use, which in turn contributes to the accumulation and persistence of resistance genes in the microbiome. This pattern of cumulative resistome expansion is directly supported by the findings of this review, where many of the identified ARGs were not directly linked to the antibiotics used in the studies. Instead, they appeared to reflect prior antibiotic exposure, indicating that these resistance genes were already present in the gut microbiome before treatment. The major background resistant genes observed in this review include beta-lactams, aminoglycosides, vancomycin, tetracyclines, and E. coli intrinsic genes.
Clinically, the persistence and enrichment of ARGs within the gut microbiome have serious implications. Individuals may carry multidrug-resistant organisms (MDRO) such as extended-spectrum beta-lactamase (ESBL)-producing Enterobacteriaceae or vancomycin-resistant Enterococcus (VRE)66,67 even before starting antibiotic treatment, increasing the risk of difficult-to-treat infections. These resistant gut colonisers serve as reservoirs that can cause bloodstream infections, recurrent gastrointestinal infections, or translocate causing sepsis, particularly in vulnerable populations such as children and immunocompromised patients.68,69 The gut microbiome’s role as a reservoir for ARGs facilitates horizontal gene transfer, further spreading resistance within and beyond the gut ecosystem. This dynamic complicates treatment options and has been linked to increased morbidity, prolonged hospital stays, and higher healthcare costs. 15
Building upon the clinical importance of ARG persistence, the detection of mdtEFOP, acrDEF and emrKY multidrug efflux pump genes in Escherichia coli carries important implications given their role in sustaining broad-spectrum antibiotic resistance.70,71 Their presence, especially in acutely ill paediatric populations as observed in this review, suggests that antibiotic use may select for highly resilient resistance mechanisms at an early age, potentially shaping long-term AMR risks. The recovery patterns reported across studies indicate that while these genes may eventually decline, they can persist for extended periods post-treatment, creating a window during which resistant bacteria may spread within individuals or communities. At the same time, signs of eventual recovery show that the microbiome can adapt, meaning resistance is not permanent but possibly influenced by the type of antibiotic used, duration of exposure, or the individual’s health status. This points to the need for cautious antibiotic use and long-term tracking of resistance genes to better manage AMR risks.
The consequences of antibiotic treatment for the gut microbiome varied considerably, depending largely on whether the host was healthy, acutely ill, or managing a chronic illness, though exposure consistently amplified the resistome across all groups. While this review focuses on antibiotic-induced disruption, it is important to note that the underlying health condition itself can perturb the gut microbiome.72–74 This creates a potential double insult where the combined effects of disease and antibiotic treatment may lead to more severe and persistent dysbiosis and resistome expansion than either factor alone.
In healthy individuals, antibiotics caused clear reductions in alpha and beta diversity in adults, and more extensive losses (including gamma diversity) in children. This suggests that even in the absence of disease, antibiotics significantly disrupt ecological balance. The resistome expands in both groups, confirming that resistance selection occurs independently of health status.
In acutely ill individuals, the adult microbiome showed greater stability in alpha diversity, possibly due to the underlying infection already altering microbial communities. Nevertheless, composition changes and increased ARGs still occur. Children experiencing acute illness exhibited broad diversity reductions and a pronounced rise in ARGs, indicating that illness may compound the disruptive effects of antibiotics.
For those with chronic illness, adults displayed typical diversity declines, while children showed unexpected stability in alpha diversity. This may point to a pre-existing, adapted microbiome in chronically ill children that is less susceptible to diversity loss. However, this stability does not extend to the resistome, which increased in all scenarios.
Gut microbial communities are central to key host functions, including immune system development and regulation of metabolic pathways.55,75,76 Our findings, however, indicate that antibiotic use can disrupt these functions. Across both adult and paediatric populations, most studies reported alterations in functional metabolic pathways, while one study identified immune dysregulation as a notable outcome. These disruptions raise concern about potential long-term health consequences, particularly in resource-limited settings where antibiotic stewardship is critical. Functional perturbations of the microbiome have been linked to a range of chronic conditions, including metabolic diseases (e.g. obesity, diabetes), gastrointestinal disorders (e.g. IBS), neurodegenerative diseases (e.g. Parkinson’s, Alzheimer’s), autoimmune diseases (e.g. ulcerative colitis), and even cancer (e.g. colorectal, pancreatic).15,77,78 These associations emphasise the need to consider not only microbial composition but also the broader functional consequences of antibiotic use for human health.
A child-focused study reported that maternal antibiotic exposure from parturition through the early neonatal period resulted in decreased alpha diversity, significant changes in beta diversity, and notable shifts in taxa composition. 41 Although the neonates themselves were antibiotic-naïve, maternal antibiotic treatment prior to birth led to notable disruptions in their gut microbiome along with functional changes (neonatal infection and inflammation). This finding is supported by a systematic review and meta-analysis by Grech et al., 79 which identified maternal antibiotic exposure as a key factor directly influencing the infant gut microbiome. Additionally, Lim et al. 80 showed that the gut microbiome is rapidly acquired after birth, a process further influenced by mode of delivery. Vaginal delivery has a greater impact on the gut microbiome compared to caesarean section,15,58 a point reinforced by Zhang et al., 81 who observed that approximately 74% of maternal microbial strains were present in vaginally delivered infants, compared to only 13% in those delivered by caesarean section.
Although the maternal-neonate study did not directly assess ARGs, the current findings, which show that maternal treatment directly alters the neonate’s microbiome, combined with established literature, suggest a strong potential for the neonate microbiome to acquire maternal microbial strains that may harbour ARGs. This transfer could render critical therapeutic agents ineffective from a very early stage of life. Furthermore, the disruption of the neonate microbiome may have major long-term implications, including an increased risk of paediatric obesity and its associated metabolic disorders, which represent growing global health challenges.82,83 Such dysbiosis can lead to endotoxin-induced low-grade inflammation (a key factor in insulin resistance) as well as altered energy absorption and bile acid metabolism. 82
Future research should prioritise investigating maternal antibiotic exposure, particularly in developing countries, as it may significantly shape the early infant microbiome and introduce prevalent ARGs that compromise treatment efficacy during childhood.
Finally, results from this review indicate that microbiome and resistome recoveries are possible, but not in all cases. This is evident in some studies that engaged in longer follow-up durations to assess recovery potentials and found the microbiome to recover after a longer period than a shorter period of observation. The resistome still contained prevalent ARGs even after longer periods of observation, suggesting no recovery. In terms of comparative recovery between populations, adults were found to have higher microbiome recovery than children, again, confirming the resilience of an adult gut microbiome. Notably, recovery outcomes are also possibly influenced by health status. Chronically ill adults demonstrated full microbiome recovery in one study, whereas healthy and acutely ill children frequently showed no recovery. This implies that baseline health mediates not only the initial disruption but also the capacity for restoration, with chronically ill hosts sometimes exhibiting altered resilience patterns. Nevertheless, the persistence of ARGs was common across all health states, emphasising that resistome expansion is a universal consequence of antibiotic treatment, regardless of the host’s health.
Respiratory microbiome and resistome
In the respiratory microbiome of children, differences were found in the alpha and beta diversities of the two included studies. As one study reported a decrease in alpha diversity, the other reported stability. In terms of beta diversity analysis, one study reported significant differences between treatment arms, while the other reported no significant differences. These contrasting results could not give a general overview of the microbial diversity. This is largely due to the fact that this particular environment is largely understudied in terms of antibiotic exposure assessment, and hence, general conclusions could not be drawn on respiratory microbial diversity. However, two results were consistent between the two studies, one being a significant change in taxa composition after exposure, and the other being an increase in ARGs. One study reported on the functional changes associated with antibiotic exposure, in which an altered functional metabolic pathway was observed. Additionally, one study found the respiratory microbiome to be fully recovered after follow-up.
Limitations
This review has several limitations worth noting. A primary challenge was the insufficient number of studies focusing on the adult gut microbiome, which restricted our ability to make strong comparisons with paediatric populations. Furthermore, the functional consequences of antibiotic exposure were severely underreported across the included studies (two adult and five child studies). This gap is not due to methodological limitations but rather a predominant focus of the primary literature on taxonomic and resistome profiling, with most studies not designed to assess functional metabolic changes via metagenomic or meta-transcriptomic analysis.
Additionally, the limited availability of research on the respiratory microbiome impeded efforts to perform thorough comparisons or draw reliable conclusions in that area. The predominance of respiratory microbiome studies conducted in children likely reflects the critical importance of early-life airway microbial colonisation. Overall, this review could not conduct a robust general comparison between the gut and respiratory microbiomes due to these significant disparities in available evidence.
Conclusion
Antibiotic use significantly disrupts the gut and respiratory microbiomes, driving compositional changes, expansion of resistance genes, and functional shifts with potential long-term health implications. Children are especially vulnerable, showing reduced recovery potential compared to adults, while the respiratory microbiome remains critically understudied despite evident susceptibility to antibiotic pressure.
Our findings emphasise the urgent need to strengthen antibiotic stewardship programs in LMICs, where rising antibiotic use and weak oversight accelerate resistance. Practical strategies include paediatric-focused prescribing guidelines that limit broad-spectrum use, investment in affordable rapid diagnostics to guide therapy, integration of stewardship into maternal-child health to safeguard neonatal microbiomes, and expansion of community-level resistance surveillance to inform empirical treatment. These context-specific interventions are vital to preserving antibiotic efficacy and mitigating long-term risks.
Future research should address key gaps by conducting longitudinal studies on resistome persistence and recovery, particularly in children; examining the impact of maternal antibiotic exposure on neonatal microbiome development; characterising the airway resistome and its clinical consequences; and testing microbiome-restorative interventions such as probiotics or prebiotics alongside antibiotics. Addressing these priorities will strengthen both clinical outcomes and stewardship strategies in LMICs.
Supplemental Material
sj-docx-1-tai-10.1177_20499361251389738 – Supplemental material for Antibiotic impact on human microecology in low- and middle-income countries: a systematic age-stratified review of gut and respiratory microbiome and resistome
Supplemental material, sj-docx-1-tai-10.1177_20499361251389738 for Antibiotic impact on human microecology in low- and middle-income countries: a systematic age-stratified review of gut and respiratory microbiome and resistome by Samuel Nee-Amugie Yartey, Aaron Awere-Duodu, Anastasia Akosua Asantewaa and Eric S. Donkor in Therapeutic Advances in Infectious Disease
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.